Microscopy Images
Cystinosis is a rare autosomal recessive lysosomal transport disorder with an incidence of 1 in 100,000 to 200,000 live births. It is the most common cause of Fanconi syndrome in children and presents clinically in 3 different forms, including infantile, juvenile (late onset), and adult (benign). Children with infantile nephropathic cystinosis have early onset of polyuria, polydipsia, dehydration, and Fanconi syndrome by the first decade of life, with progressive decreases in kidney function, reaching end-stage renal disease (ESRD) by a mean age of 9 years. Systemic deposits of cystine also occur, resulting in end-organ damage in eyes, liver, the central nervous system, muscle, and endocrine tissues.
Children of European ancestry show a characteristic “towheaded” appearance, with very light hair and skin color due to defective melanin pigmentation. Failure to thrive and short stature also develop. In severe cases, rickets may develop due to prolonged renal tubular acidosis. Patients with the juvenile variant of cystinosis have mild nephropathy without Fanconi syndrome and slow progression of kidney disease, with ESRD at a later age (12-28 years). Those with the adult form of cystinosis have ocular involvement without kidney disease (nonnephropathic).
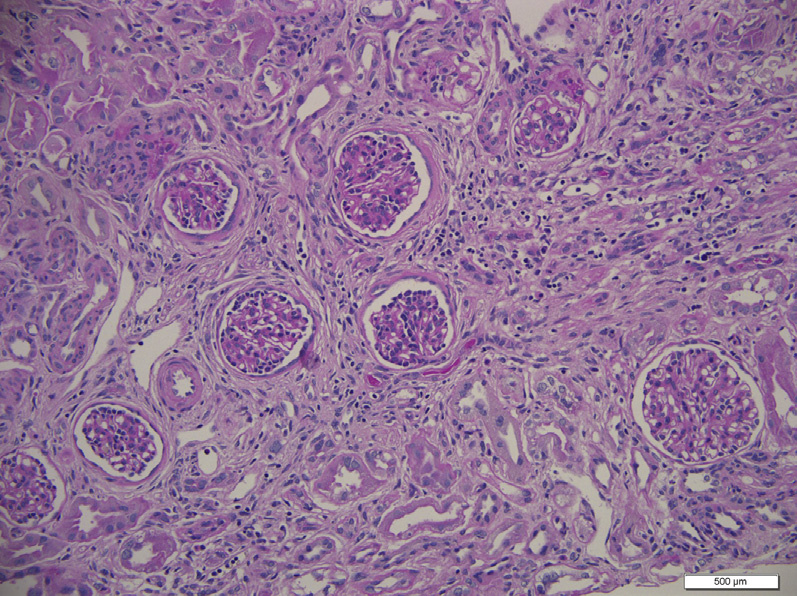
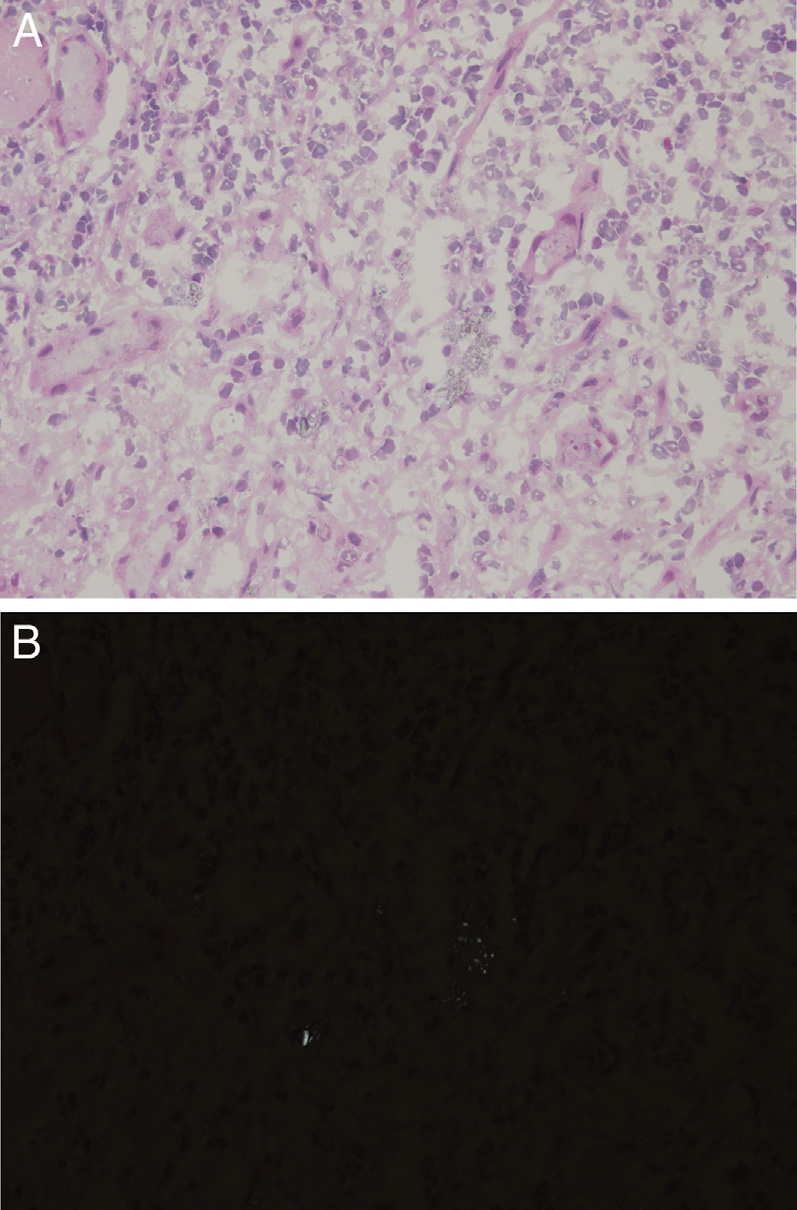
Light microscopy: Cystine crystal deposition mostly occurs in the cortical interstitium within macrophages, and can also be found in tubular and glomerular epithelial cells. Crystals are usually dissolved by routine processing and sectioning of paraffin-embedded tissues, but can be preserved with alcohol solutions. Clear spaces reflecting dissolved crystals can be detected on toluidine blue–stained electron microscopy scout slides.
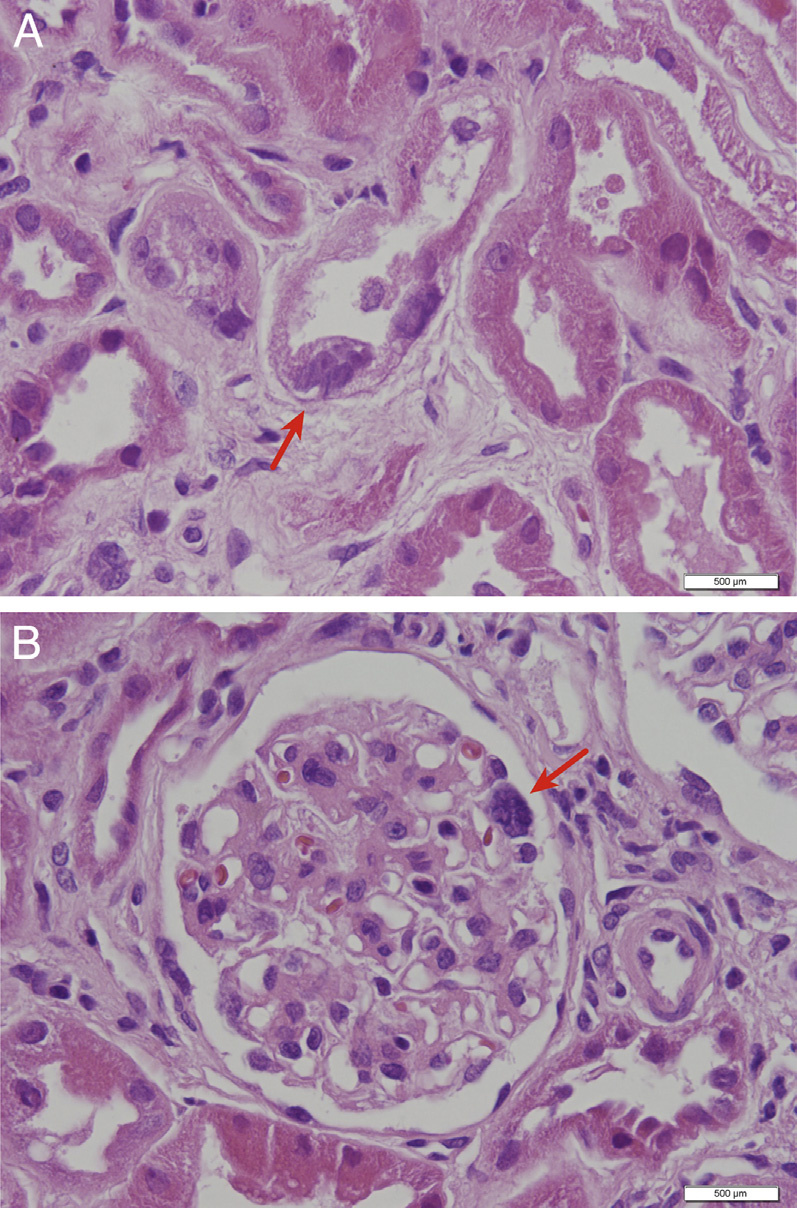
Crystals are best seen on frozen section. These crystals are hexagonal, rhombohedral, or polymorphous in shape, and birefringent under polarized light. Early morphologic changes include tubular epithelial cell flattening with loss of brush borders, and multinucleated tubular and glomerular epithelial cells, particularly involving podocytes. The first part of the proximal tubule becomes atrophic and thin, forming a “swan-neck” deformity. Ultimately, injury leads to tubulointerstitial fibrosis with progressive glomerulosclerosis.
Immunofluorescence microscopy: No specific staining.
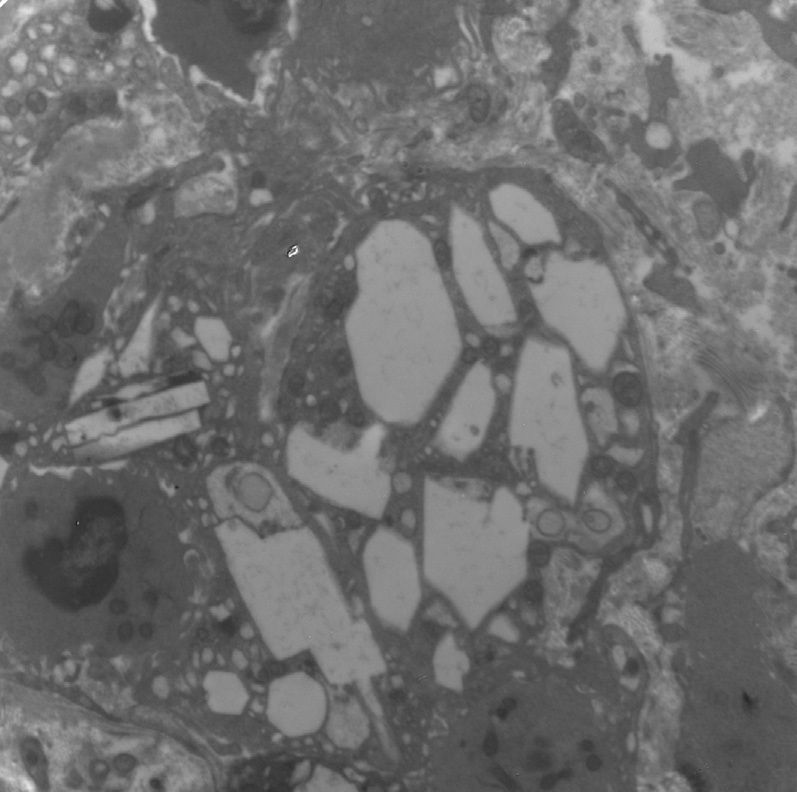
Electron microscopy: Cystine crystals are seen as clear spaces within interstitial macrophage and tubular epithelial cytoplasm. Subtotal podocyte foot process effacement may be present.
Cystinosin, a lysosomal membrane cystine transporter protein, is mutated, causing accumulation of cystine within lysosomes. This mutation maps to chromosome 17p13. In the infantile form, patients are usually heterozygous for a 57-kilobase deletion. In patients with juvenile and adult forms, a point or missense mutation involving transmembrane loops or the amino terminus of the protein is involved. Cysteamine treatment binds intralysosomal cystine, allowing excretion of cystine from lysosomes to the cytoplasm for degradation. Treatment slows disease progression.
Cystinosis recurs posttransplantation, with ongoing cystine crystal accumulation in the allograft.
Reflux/chronic pyelonephritis has a patchy, well-demarcated pattern of fibrosis often seen in cystinosis; however, thyroidization of tubules and the absence of cystine crystals distinguish it from cystinosis. Oxalosis has intratubular, clear, fan-shaped crystals that are birefringent under polarized light.
Cystinuria presents with recurrent nephrolithiasis without cytoplasmic cystine crystal accumulation, and without multinucleated podocytes, and does not recur in the allograft. Fabry nephropathy shows vacuolization of podocytes by light microscopy and characteristic multilamellated myelin figures and zebra bodies within podocytes and other cells by electron microscopy.