Microscopy Images
Patients with Alport syndrome present with hematuria in early childhood. Proteinuria develops during teenage years, with nephrotic syndrome in 30%-40%.
Male patients with X-linked Alport progress to end-stage kidney disease, usually in adulthood. Female carriers of X-linked classic Alport syndrome usually have only hematuria, but may develop progressive kidney disease as middle-aged or older adults. Carriers of rare autosomal recessive variants of Alport syndrome also have hematuria.
Gradual deafness develops in about half of adult men with Alport syndrome, with eye defects in about onethird, most commonly anterior lenticonus. Alport syndrome patients who receive a transplant may develop anti–glomerular basement membrane (GBM) antibodies to the normal type IV collagen in the transplant, with resulting crescents and anti-GBM antibody–mediated glomerulonephritis occurring in 2% to 5%.
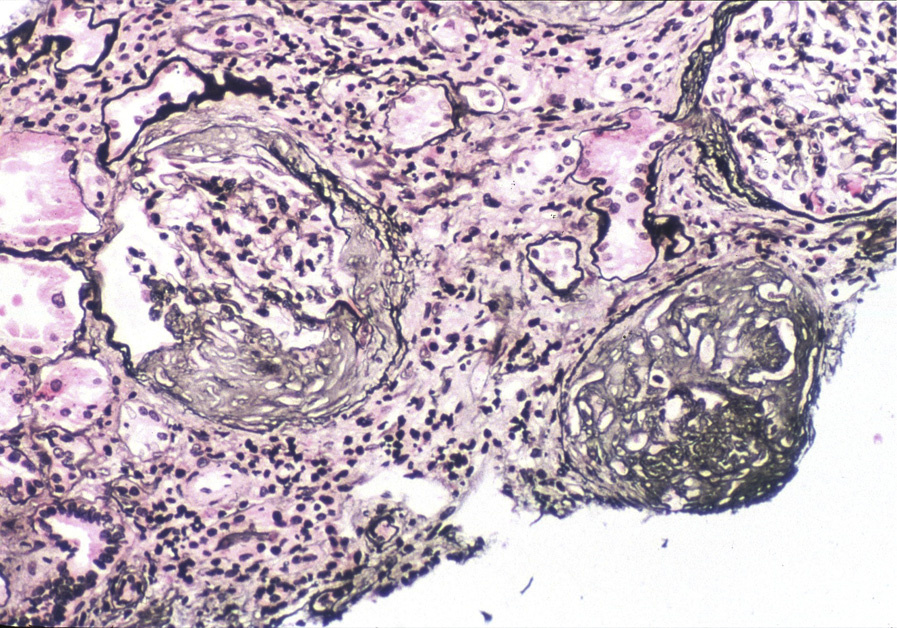
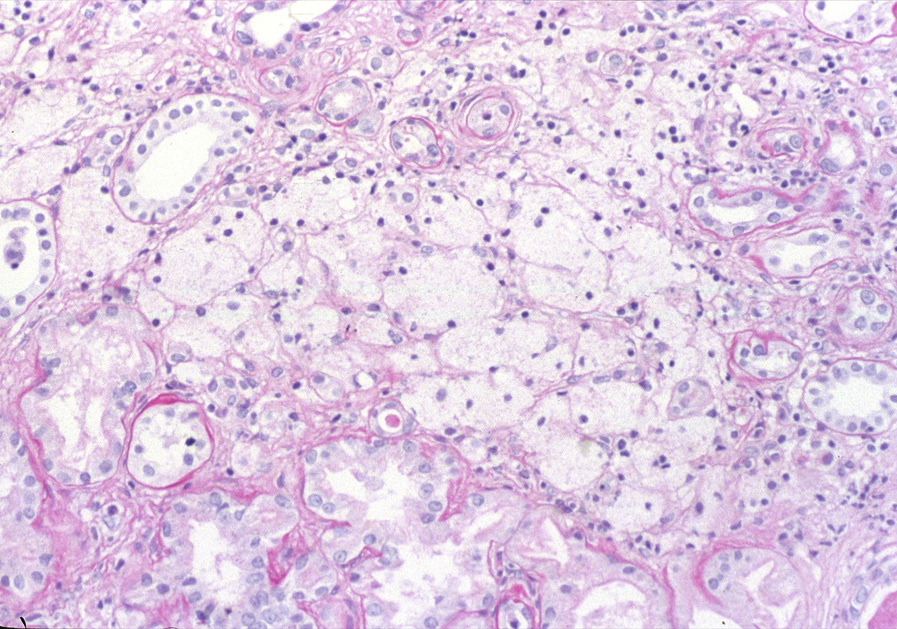
Light microscopy: Kidney biopsy is unremarkable in early stages in males with X-linked Alport syndrome and in carriers, with afflicted patients showing later secondary glomerulosclerosis, interstitial fibrosis, and prominent interstitial foam cells, the latter a response to longstanding proteinuria.
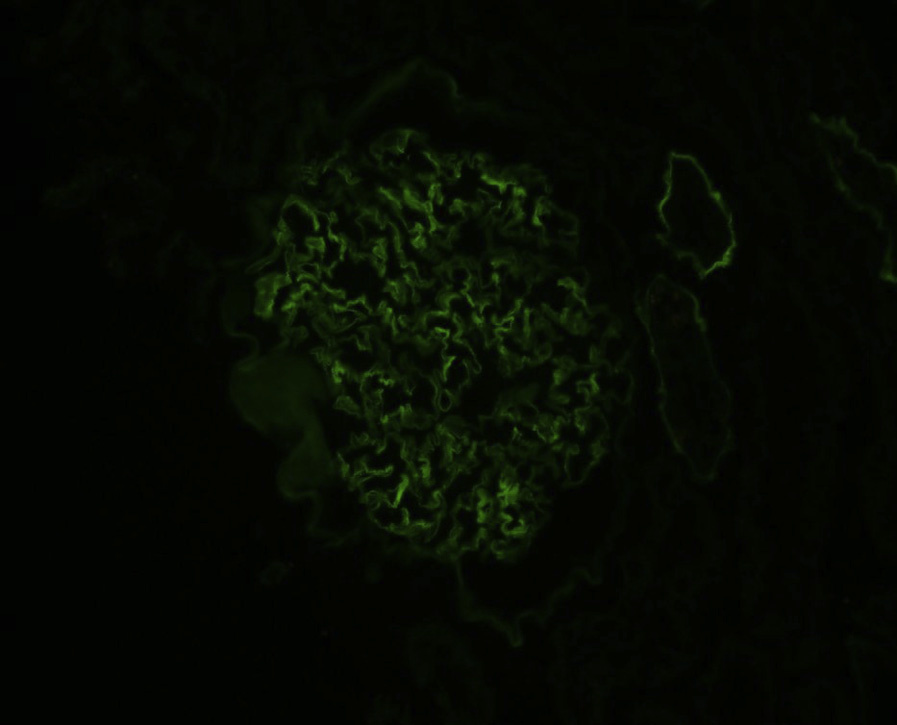
Immunofluorescence microscopy: Standard immunofluorescence shows no immune complexes.
Immunostaining for type IV collagen alpha chains can confirm absence of alpha 5 type IV collagen in X-linked Alport patients, mosaic patchy staining in carrier females, and presence of alpha 5 type IV collagen only in Bowman’s capsule and distal tubules but not in GBMs in patients with autosomal recessive Alport. Carriers of autosomal recessive Alport have normal immunostaining for type IV collagen alpha chains. Of note, a subset of patients with Alport still have residual truncated type IV collagen chain and may show normal immunostaining.
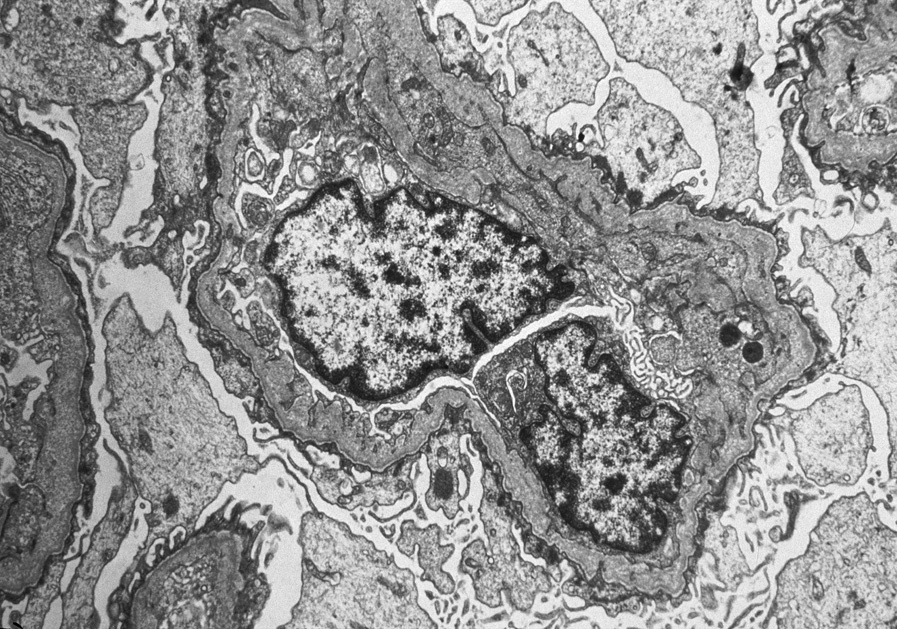
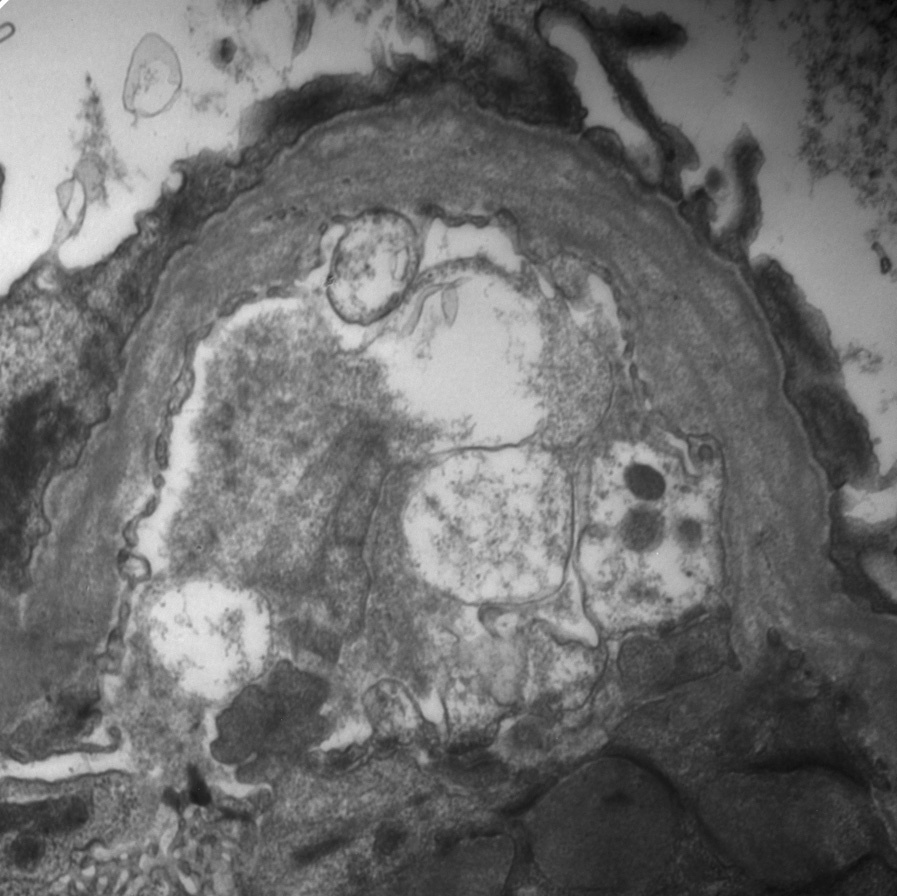
Electron microscopy: Early Alport or carriers of Alport show diffuse thinning of GBMs. In established Alport, there are irregular thin and thickened areas with splintered and irregular multi-laminated appearance of the lamina densa, with short stubs of fibrils at right angles to the GBM, irregular lucent areas interspersed with thickened areas of the lamina densa, resulting in a so-called basket-weave pattern. There are no immune complexes, and foot processes, particularly in sclerosed glomeruli, may show effacement.
Alport syndrome is caused by mutations in alpha 3, alpha 4, or alpha 5 type IV collagen, resulting in defective assembly of the alpha 3, 4, 5 heterotrimer that is essential for function in the GBM, the lens of the eye, and the cochlea of the ear. This results in the classic clinical manifestations of established Alport syndrome.
Alport carriers have diffusely thin GBMs. Socalled benign familial hematuria frequently represents a carrier state of autosomal recessive Alport, and biopsies show thin basement membranes morphologically. Segmental thinning of GBMs may occur nonspecifically. Chronic immune complex disease with resorbed deposits resulting in areas of lucency may have similar EM appearance as thickened GBM areas in Alport, and is distinguished by IF and other EM findings. Irregular or thin glomerular basement membranes may also be seen in patients with Frasier (WT1 mutation) or Pierson (LAMB2 mutation) syndrome, both with normal type IV collagen staining.