Microscopy Images
Lecithin–cholesterol acyltransferase (LCAT) deficiency is a rare (prevalence <1:1,000,000) autosomal recessive disease.
Clinical features include corneal opacities, normochromic normocytic anemia, and proteinuria. Multiple lipoprotein abnormalities are present (see Etiology/Pathogenesis below). Corneal opacities are observed from early childhood. Renal involvement is a major cause of morbidity and mortality, starts with proteinuria in childhood, and progresses to ESRD by the 4th or 5th decade. The clinical manifestations may differ markedly depending on which of over 90 mutations are present in the LCAT gene on chromosome 16q22. Homozygous mutations may lead to familial LCAT deficiency (no enzyme activity; more severe symptoms) or fish eye disease (partial enzyme deficiency; corneal opacity is the main morbidity). Recurrence of lesions after kidney transplantation can occur early; however, allograft function often persists long term despite the presence of deposits.
Acquired LCAT deficiency can occur due to inhibitory autoantibodies against LCAT. Serum lipoprotein abnormalities and the pathologic features of acquired and congenital LCAT deficiency are similar; however, the former is commonly associated with membranous nephropathy (due to anti-LCAT antibodies), which is not a feature of the latter.
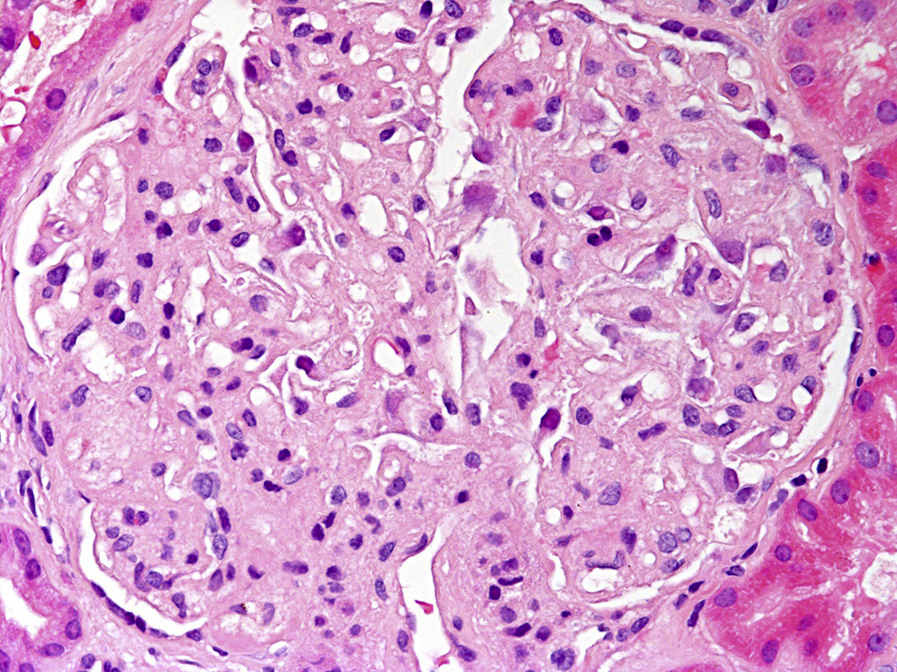
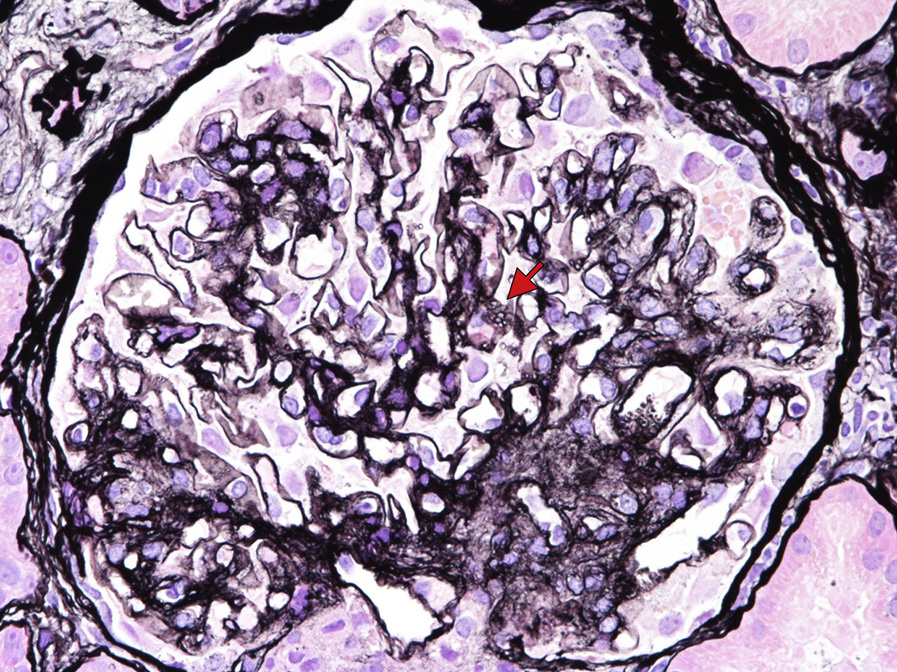
Light microscopy: Glomerular capillary walls become vacuolated and glomerular basement membranes (GBMs) may show spiculation on silver stain, mimicking membranous nephropathy. As the disease progresses, GBM duplication with a bubbly, vacuolated, and honeycomb appearance on silver and periodic acid–Schiff stains due to lipid deposition becomes more prominent.
The enlarged mesangial area shows mild vacuolation. Similar findings are also observed in the arterioles and interlobular arteries. Renal tubules become atrophic with proportional interstitial fibrosis as the disease progresses.
Immunofluorescence microscopy: Immunofluorescence studies are typically negative. C3 deposition— likely a nonspecific finding—has been reported in some patients. Acquired LCAT deficiency with concomitant membranous nephropathy shows granular IgG and C3 staining of glomerular capillary walls.
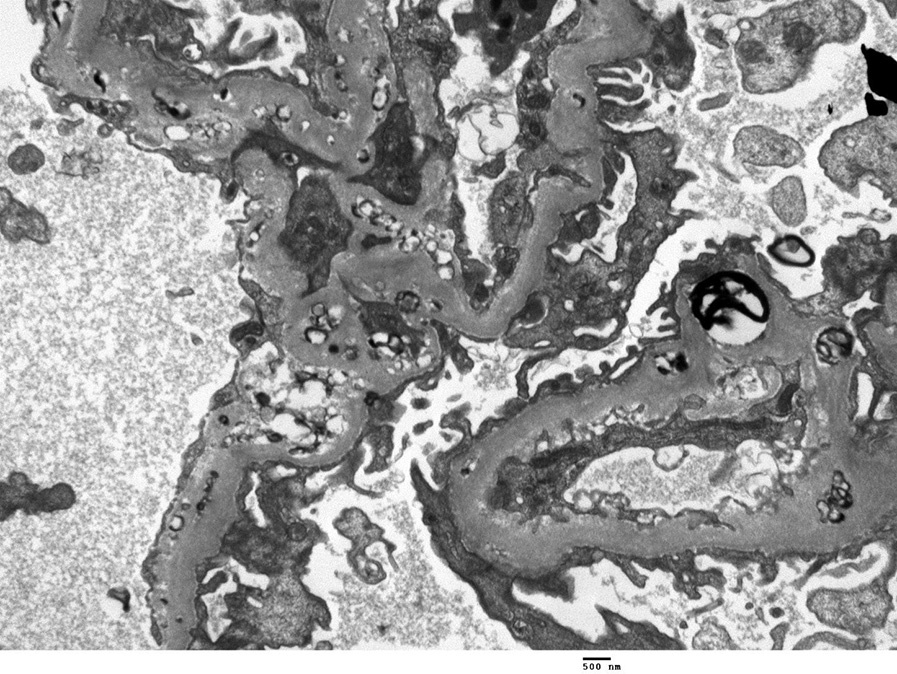
Electron microscopy: Lipid deposits appear as small dark, irregular, granular, partially electrondense/partially electron-lucent particles.
These are present in large numbers in the subepithelial, subendothelial, intramembranous, and mesangial regions. Glomerular basement duplication may be present. Similar deposition is also often found in the Bowman capsule. Electron-lucent vacuoles are present in the GBM and mesangial matrix, creating a “moth-eaten” appearance. Larger, organized lipid-containing deposits with unusual large intraluminal thrombus-like deposits with tubular substructure can rarely be present. Subepithelial immune type deposits characteristic of membranous nephropathy may accompany acquired LCAT deficiency.
LCAT is a plasma enzyme, mainly synthesized in the liver, which esterifies free cholesterol, and functions in the formation of mature HDL and reverse cholesterol transport from the peripheral tissues to the liver. LCAT deficiency is associated with lipoprotein abnormalities including low HDL-cholesterol, increased serum free cholesterol, mild to severe hypertriglyceridemia, presence of lipoprotein X (Lp-X), decreased serum apolipoprotein (apo) A-I and A-II, and increased apo E. Lp-X, a large multilamellar phospholipid vesicle containing various apolipoproteins without a neutral lipid core, is present in lipid deposits in the kidney and is likely involved in disease pathogenesis. Deposition of lipids on the red blood cell membrane leads to hemolysis and anemia.
Hepatic glomerulopathy, also called hepatic glomerulosclerosis, is a secondary sclerosing lesion associated with liver disease, and shows deposition of lipid particles, similar to those seen in LCAT deficiency, but usually with less severity.
Hepatic glomerulopathy occurs in the context of liver failure and cirrhosis. GBM changes on silver stain in LCAT deficiency may mimic membranous nephropathy stage III. Immunofluorescence and electron microscopy findings can differentiate between these two conditions. In acquired LCAT deficiency there may be concomitant membranous nephropathy; presence of lipid granules is the key to diagnosis of the underlying LCAT abnormality.
Pathologic findings of Alagille syndrome are very similar to those of LCAT deficiency, with numerous lipid particle deposits in the mesangium and along the GBM. However, the autosomal dominant pattern of inheritance, and the presence of cholestasis and cardiac, skeletal, ocular, and facial abnormalities differentiate this condition from LCAT deficiency. Nail-patella syndrome creates a moth-eaten GBM appearance, but lacks glomerular lipid deposits.