Microscopy Images
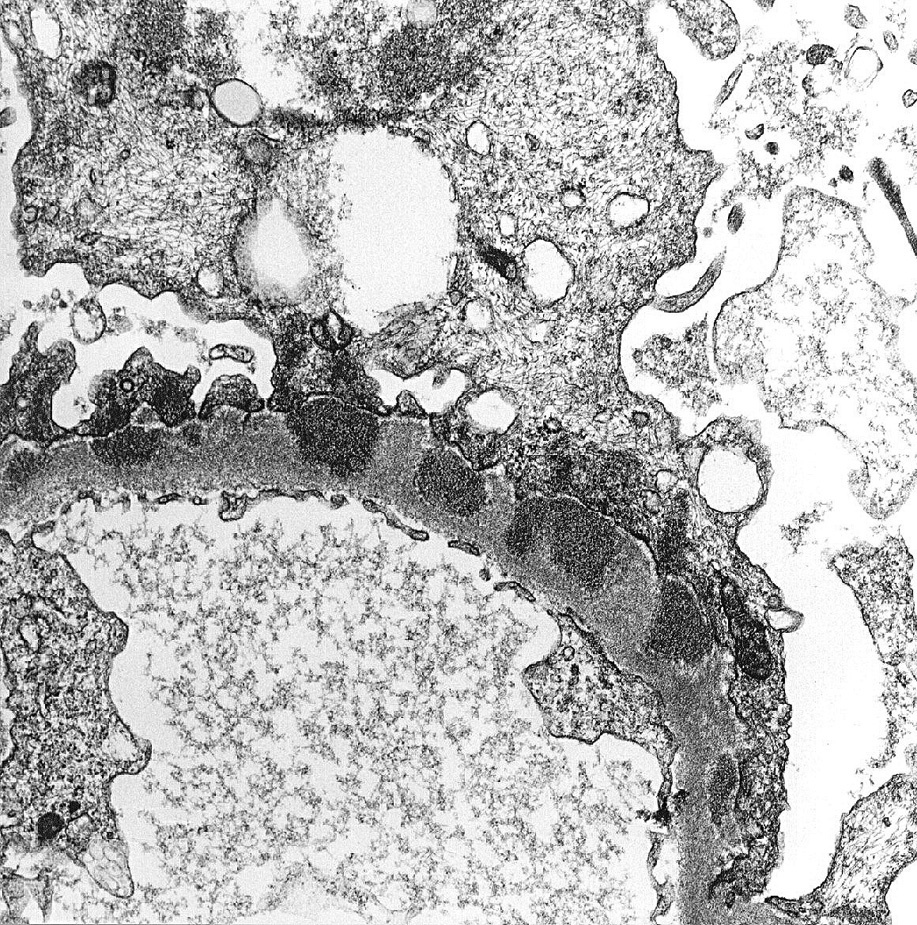
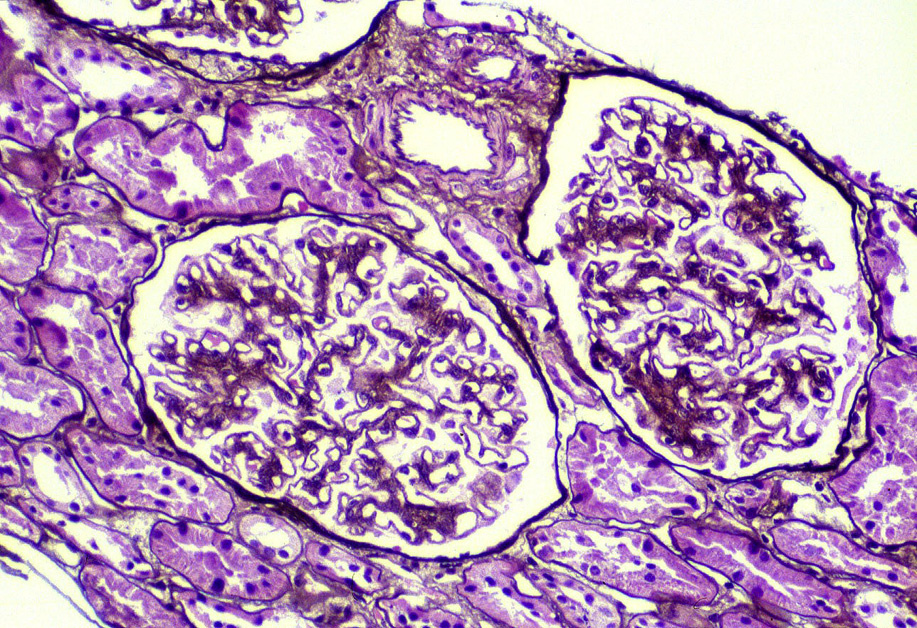
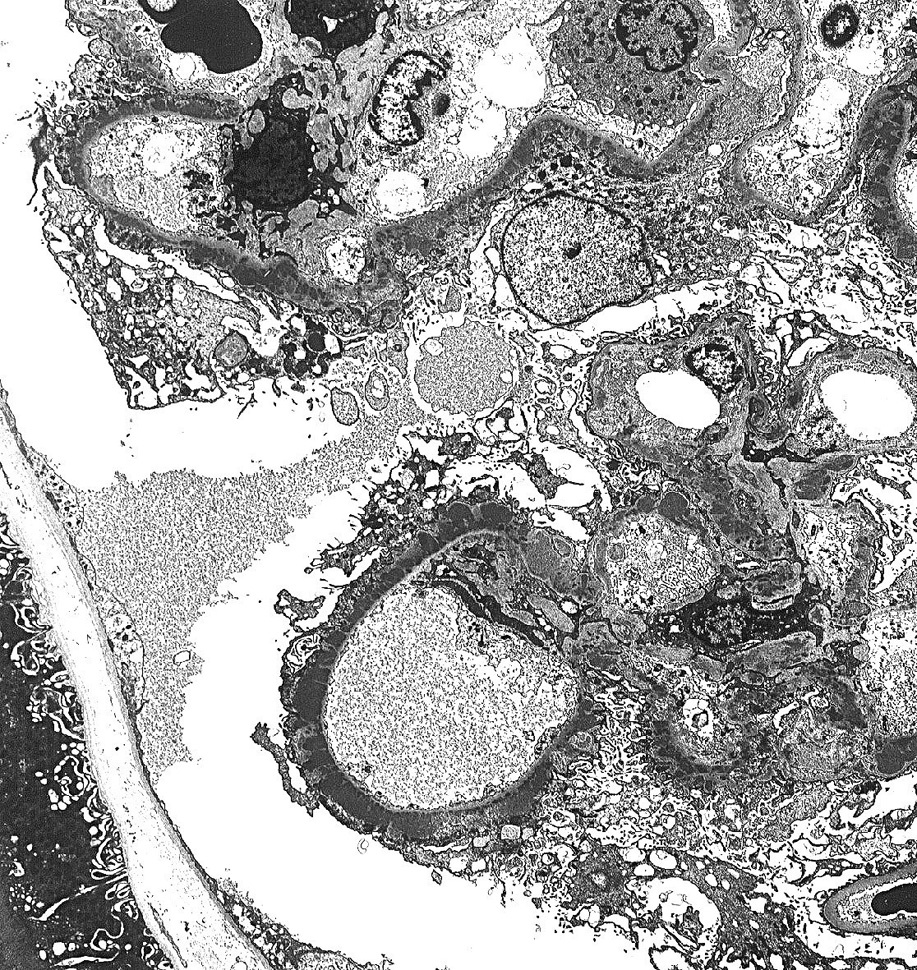
Membranous nephropathy (MN) is an immune complex disease caused by subepithelial deposits.
Primary MN is a common cause of nephrotic syndrome. About a third of patients reach remission, a third are stable, and a third have progressive loss of kidney function and persistent proteinuria.
The phospholipase A2 receptor (PLA2R), expressed on podocytes, is the antigen in about 70% of patients with primary MN. Localization of shed PLA2R within the granular deposits in the subepithelial area thus indicates high likelihood of primary MN, rather than a secondary etiology.
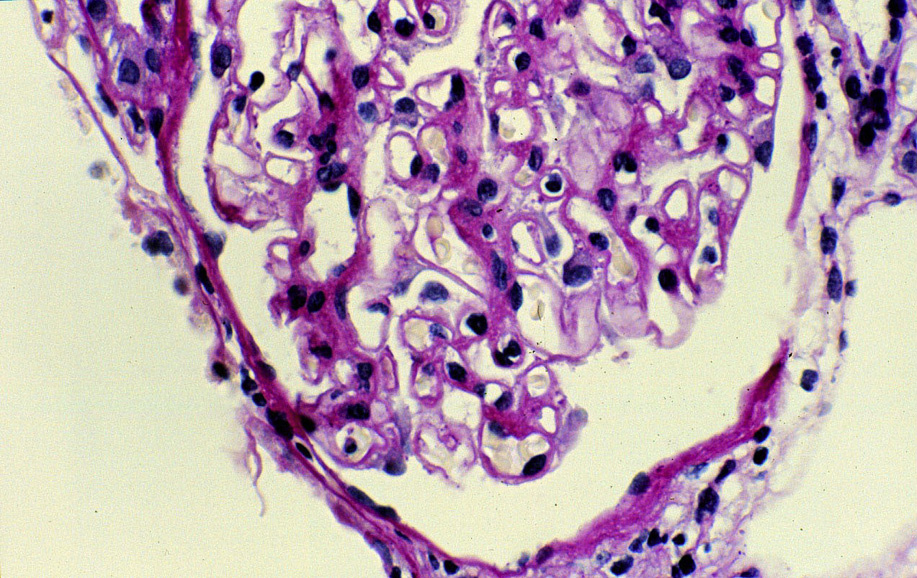
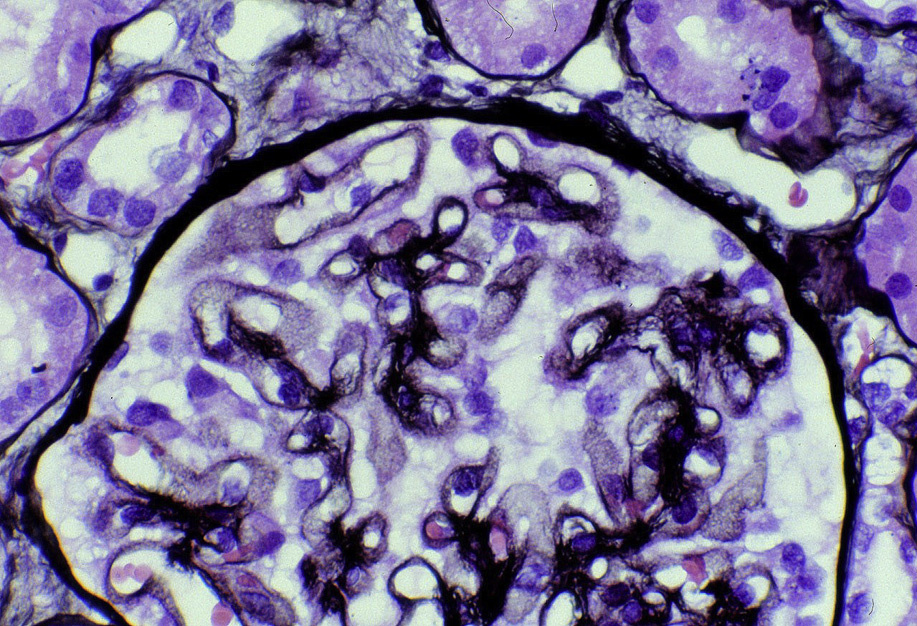
Light microscopy: The earliest stages may have no alterations visible by light microscopy. The early light microscopy lesion consists only of prominent glomerular basement membranes (stage I). In early MN, small deposits may be visualized as a pinpoint “hole” on Jones silver stain, which does not stain the deposits. Ongoing glomerular basement membrane matrix reaction to the deposits results in a “spike” appearance resulting from projections of basement membrane matrix out towards the urinary space on silver stain (stage II). Segmental sclerosis and tubulointerstitial fibrosis may ensue with progressive disease.
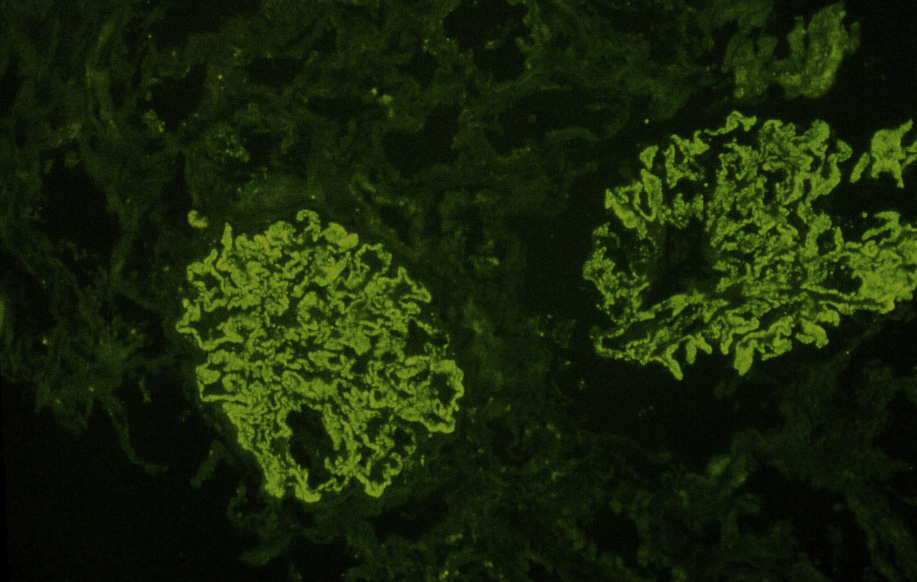
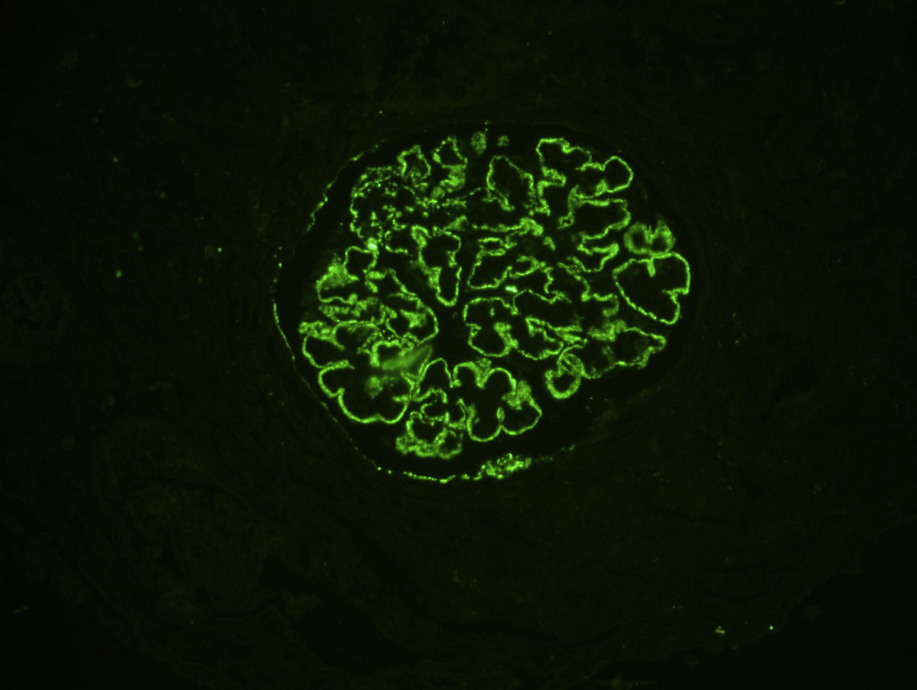
Immunofluorescence microscopy: Granular capillary wall deposits of polyclonal immunoglobulin G (IgG; often IgG4 dominant in primary MN), with variable C3, and corresponding positive granular capillary wall PLA2R staining (performed on formalinfixed pronase-digested biopsy sections).
Electron microscopy: Extensive foot process effacement and subepithelial deposits with increasing matrix spike reaction with advancing disease. This matrix reaction can ultimately envelop the deposits, resulting in a laddering appearance (stage III).
Deposits may become rarified in chronic disease (stage IV). Mesangial deposits are absent or rare in primary MN.
Primary MN is caused by autoantibodies to PLA2R in about 70% of patients. The inciting event(s) causing this autoantibody remain largely unknown.
Some patients with MN initiated by nonsteroidal anti-inflammatory drugs may also show PLA2R positivity.
Patients with primary MN usually have circulating anti-PLA2R antibodies that bind to the PLA2R on podocytes. The immune complexes formed in situ are shed into the subepithelial area, fix complement, and injure podocytes, resulting in injury, effacement, and marked proteinuria.
A membranous pattern of injury with spike formation may be seen in other diseases with capillary wall deposits. These typically are PLA2R negative, and show mesangial deposits and other distinctive features: proliferative glomerulonephritis with monoclonal deposits (diagnosed by the clonal staining with only κ or only λ light chains); amyloid or fibrillary glomerulonephritis (diagnosed by characteristic fibrils shown by electron microscopy); membranous lupus nephritis (“full-house” staining, ie, with IgG, IgA, IgM, C3, and C1q, mesangial deposits, and reticular aggregates); or other secondary causes of MN.