Microscopy Images
Pierson syndrome is an autosomal recessive disease that accounts for about 2.5% of nephrotic syndrome within the first year of life, usually manifested clinically within the first 3 months. Patients typically present with massive proteinuria and edema. Renal manifestations are accompanied by neurodevelopmental abnormalities, such as hypotonia, muscular weakness/myasthenia, and ophthalmic findings, most often microcoria. Glaucoma, cataracts, and retinal detachment can also occur. Patients with the most severe manifestations die within the first year of life, while those with less severe manifestations progress to chronic kidney failure by 10 years of age and then require transplantation. Patients can also have limited Pierson syndrome, typically due to a missense mutation, with limited, or rarely absent, neurologic or ophthalmic abnormalities. These patients develop nephrotic syndrome later in childhood, but still in the first decade of life.
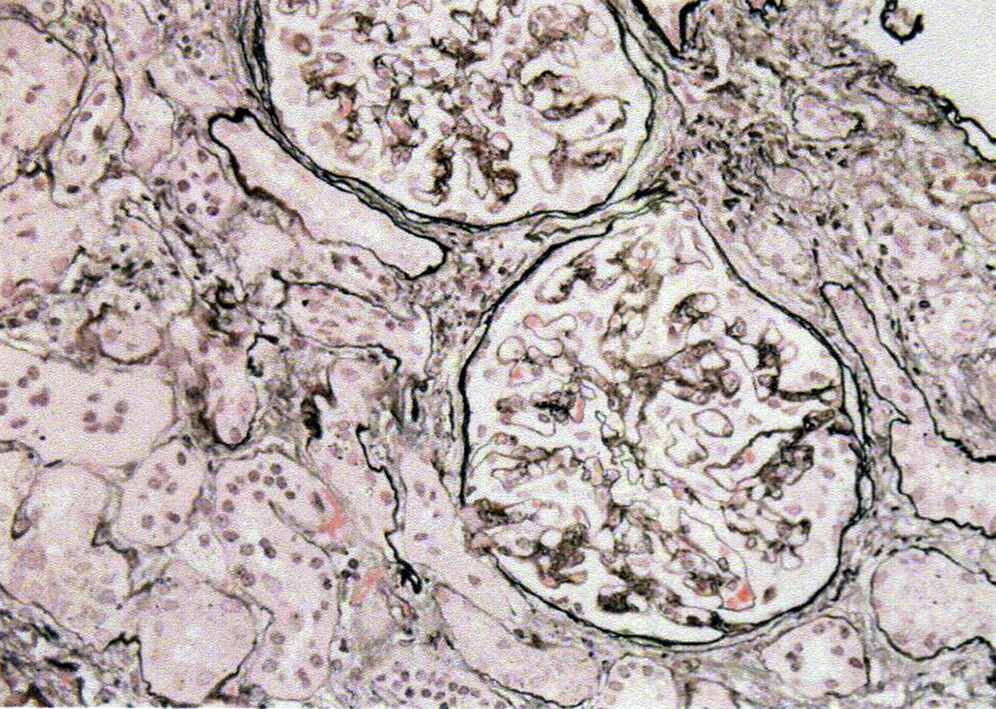
Light microscopy: Glomeruli show increased mesangial matrix without mesangial proliferation and increased global sclerosis. Podocytes may show an immature cuboidal fetal appearance. Glomerulocystic changes may be present. Crescents/pseudocrescents have been described in some patients, but without necrosis or glomerular basement membrane (GBM) breaks. There is proportional tubulointerstitial atrophy and fibrosis.
Immunofluorescence microscopy: There are no immune complexes by routine immunofluorescence staining. Additional research studies can document absence of the laminin β2 chain in classic Pierson syndrome, with persistent but decreased staining in some patients with renal-limited Pierson syndrome.
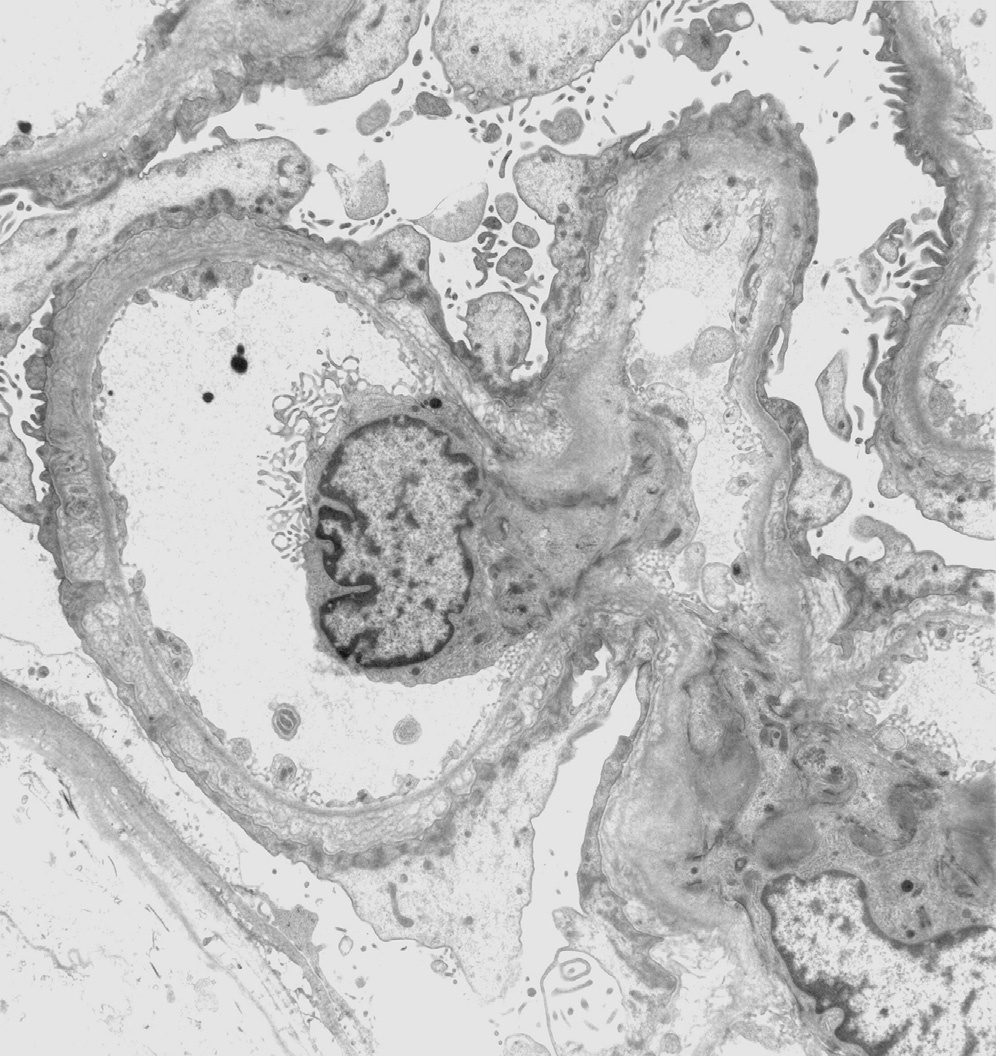
Electron microscopy: There is increased mesangial matrix without deposits. Podocytes show extensive foot process effacement with irregular thick and thin zones of the GBM both on the subepithelial as well as subendothelial aspects of the lamina densa. In addition, the lamina densa may show lamellation and corrugation.
Pierson syndrome is an autosomal recessive disease due to mutation in LAMB2 (3p21), which encodes the laminin β2 chain. This laminin subunit is an important component of the glomerular and ocular basement membranes, retina, and neuromuscular junctions. A truncating mutation causes more severe clinical disease, while a missense mutation presents with milder and often delayed clinical disease.
The differential diagnosis includes other causes of congenital/early nephrotic syndromes, some with abnormal GBM by electron microscopy. They are distinguished by age of onset, clinical findings, and distinct morphologies as follows. Denys-Drash syndrome presents with nephrotic syndrome usually before age 2 with male gonadal dysgenesis and mutations in exons 8 or 9 of WT1, with risk for Wilms tumor and gonadoblastoma. Kidney biopsy shows diffuse mesangial sclerosis and GBMs may be irregularly thickened with multilayering by electron microscopy. Frasier syndrome usually presents with nephrotic syndrome at age 2 to 6 years and is associated with pseudohermaphroditism with external female genitalia with streak gonads, XY karyotype, and mutations of intron 9 of WT1. Kidney biopsy shows focal segmental glomerulosclerosis (FSGS) and GBM irregular thickening with foot process effacement.
Galloway-Mowat syndrome presents with nephrotic syndrome within the first month of life and is associated with microcephaly and seizures. Kidney biopsy can show diffuse mesangial sclerosis or FSGS. Electron microscopy shows GBM irregularity with expansion of lamina rara interna and lamina rara externa with foot process effacement. The genetic basis for this syndrome is unknown.
Congenital nephrotic syndrome of the Finnish type is due to mutations in nephrin (NPHS1). Kidney biopsy shows no specific glomerular changes by light microscopy with cystic dilation of tubules. There is extensive podocyte foot process effacement with absent slit diaphragms by electron microscopy, but GBMs appear normal. Multiple types of genetic mutations involving NPHS1 can be causative.