Microscopy Images
The nail-patella syndrome (OMIM #161200) or hereditary osteo-onychodysplasia (HOOD syndrome) is a rare autosomal dominant disorder caused by LMX1B mutations, characterized by limb and pelvic skeletal abnormalities (eg, hypoplastic or absent patella, elbow dysplasia, and iliac horns), dystrophic nails and distal digital abnormalities, and kidney disease, with an estimated incidence of 1/50,000.
Renal involvement is the major determinant of prognosis. Besides kidney disease and skeletal and nail abnormalities, other findings may include sensorineural hearing loss, glaucoma, gastrointestinal involvement, and vasomotor dysfunction. About 10%-40% of patients develop kidney disease, manifesting with moderately increased albuminuria and proteinuria, which may present at any age or be intermittent, and is more prevalent during pregnancy.
Some patients may develop nephrotic syndrome.
Severity of renal involvement is variable within and between families. Microscopic hematuria is also common. Progression to end-stage renal disease is rare, but can occur even in early childhood. Some patients with LMX1B mutation may mainly present with a renal phenotype without skeletal or other extrarenal manifestations, a condition known as nailpatella–like renal disease (OMIM #256020) or LMX1B-associated nephropathy. Some patients with LMX1B-associated nephropathy may present as familial focal segmental glomerulosclerosis (FSGS) without showing characteristic electron microscopy findings of nail-patella syndrome. Although identification of LMX1B mutation by genotyping is an important component of nail-patella syndrome diagnosis, the lack of mutation or deletion in this gene does not exclude the diagnosis since typical features of the syndrome can rarely be seen in patients in whom such genomic alterations cannot be detected.
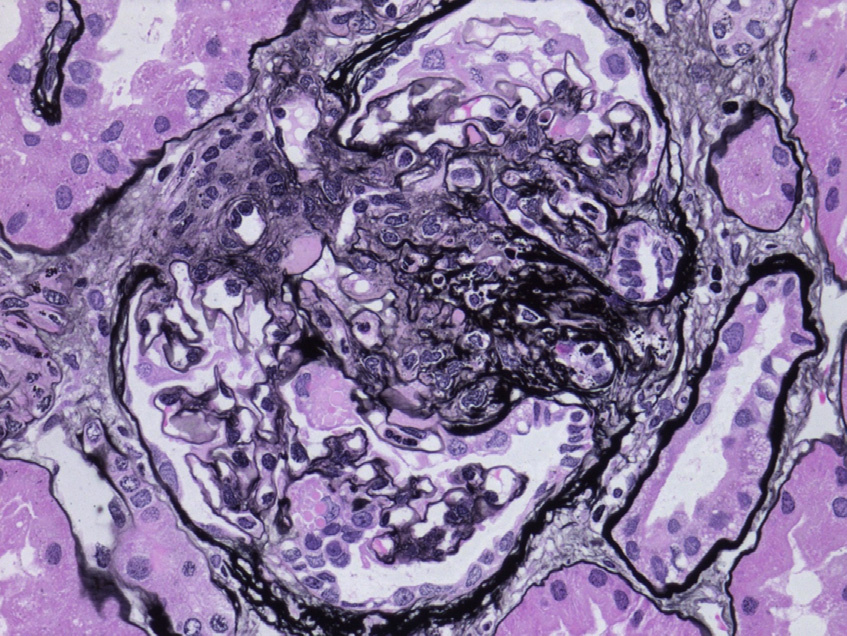
Light microscopy: Glomeruli are usually unremarkable by light microscopy. Secondary FSGS, global glomerulosclerosis, and associated tubulointerstitial fibrosis may be seen as chronicity progresses.
Immunofluorescence microscopy: Immunofluorescence studies are typically negative. Nonspecific IgM and C3 deposition may be seen in sclerotic glomeruli.
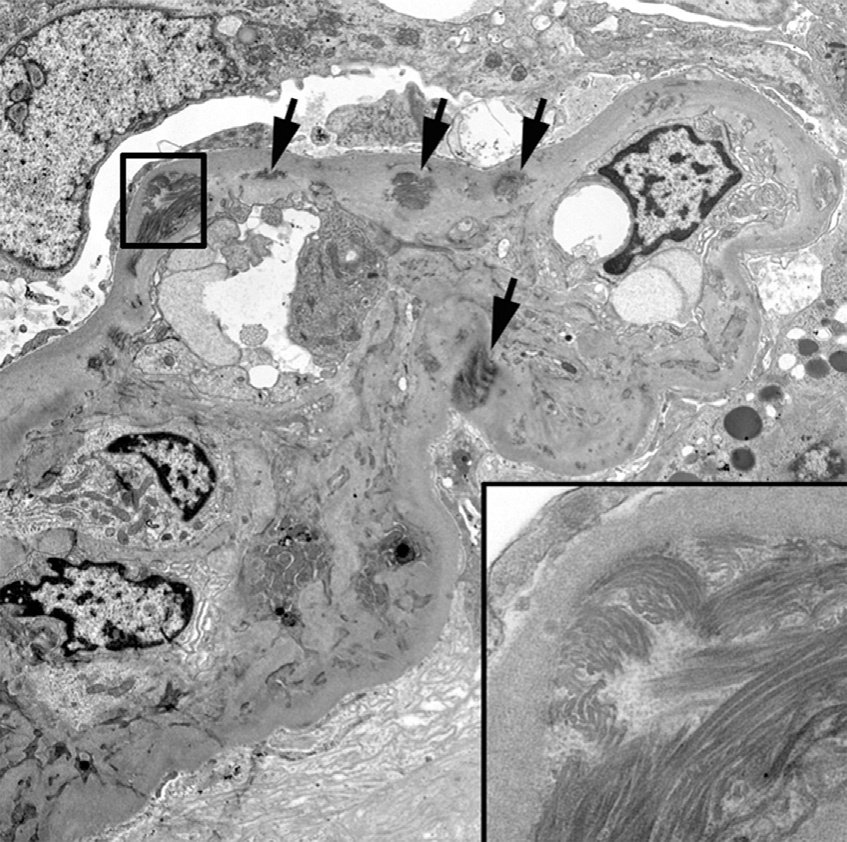
Electron microscopy: Glomerular basement membranes (GBMs) show focal or diffuse irregular thickening with electron-lucent areas (moth-eaten appearance) containing type III collagen bundles with a periodicity of about 65 nm. Similar collagen fibrils can be seen in mesangial matrix. Collagen fibril clusters may be visible by routine uranyl acetate/lead citrate staining or after enhanced staining by phosphotungstic acid or tannic acid. Podocytes show segmental effacement of foot processes. There is no correlation between the severity of the ultrastructural lesions and the clinical renal manifestations. Thus, typical electron microscopy findings may be present in the absence of clinical evidence of renal involvement.
The LMX1B gene is located at 9q34 and encodes LMX1B protein, a LIM-homeodomain transcription factor that regulates target gene expression in kidney, developing limbs, and other organs. LMX1B is important for establishing normal dorso-ventral patterning in the limbs, for differentiation of the anterior portions of the eyes, and for neuronal development in the central nervous system. In addition, LMX1B is expressed in podocytes and targets regulatory regions of the COL4A4, CD2AP, and NPHS2 (podocin) genes. Also, ABRA and ARL4C, genes for actin cytoskeleton–associated proteins, are thought to be possible targets of LMX1B.
Collagenofibrotic glomerulopathy presents with abundant accumulation of collagen type III fibrils in the glomeruli. However, the majority of the fibrils are distributed in subendothelial and mesangial regions, as opposed to nail-patella syndrome, where collagen fibrils are typically within the GBM as well as in the mesangial regions and associated with a moth-eaten appearance. Glomeruli in collagenofibrotic glomerulopathy show prominent mesangial expansion and skeletal abnormalities are absent. Glomerulopathy of hereditary multiple exostosis is characterized by diffuse mesangial and GBM fibrillar collagen deposition, in contrast to nail-patella syndrome where the fibrils show an irregular and clustered distribution.
Presence of EXT1 or EXT2 mutation and osteochondromas help in diagnosing this condition. Fibronectin glomerulopathy shows granular and focal fibrillar deposits (9-16 nm in diameter) in the glomeruli, which stain intensely for fibronectin, with nodular glomerulosclerosis by light microscopy.