Microscopy Images
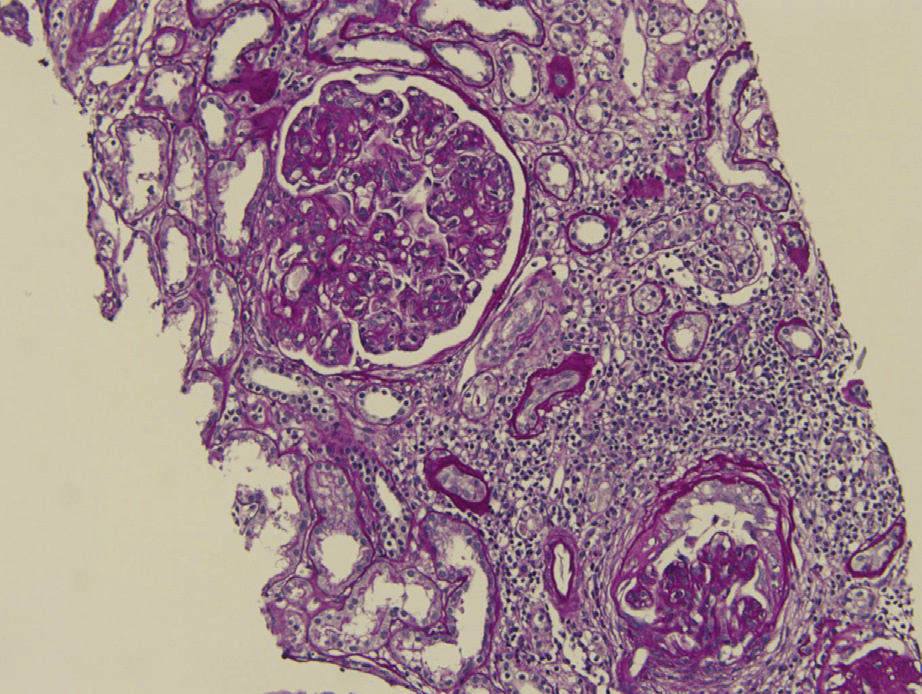
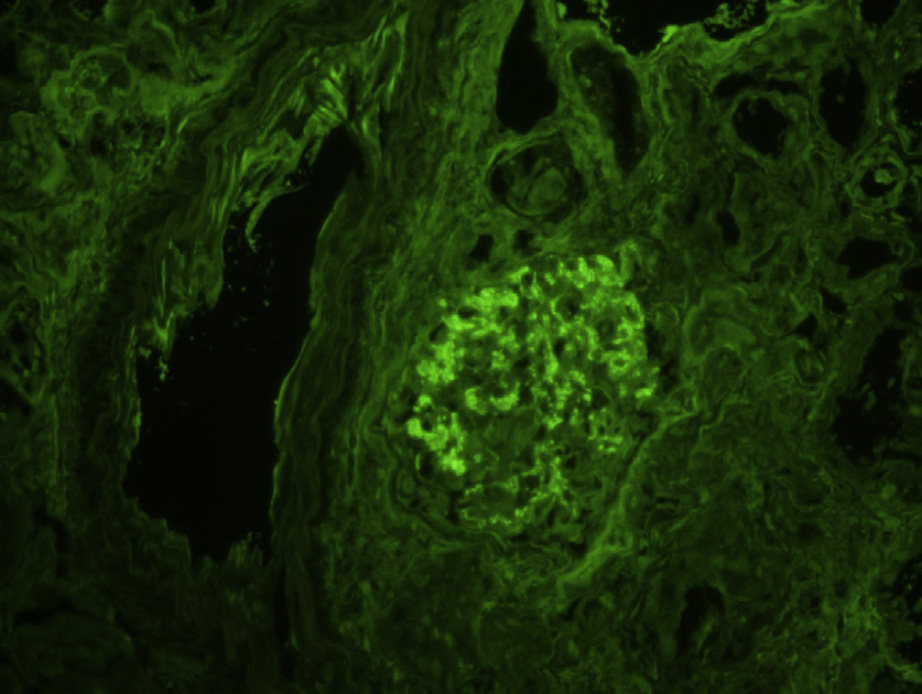
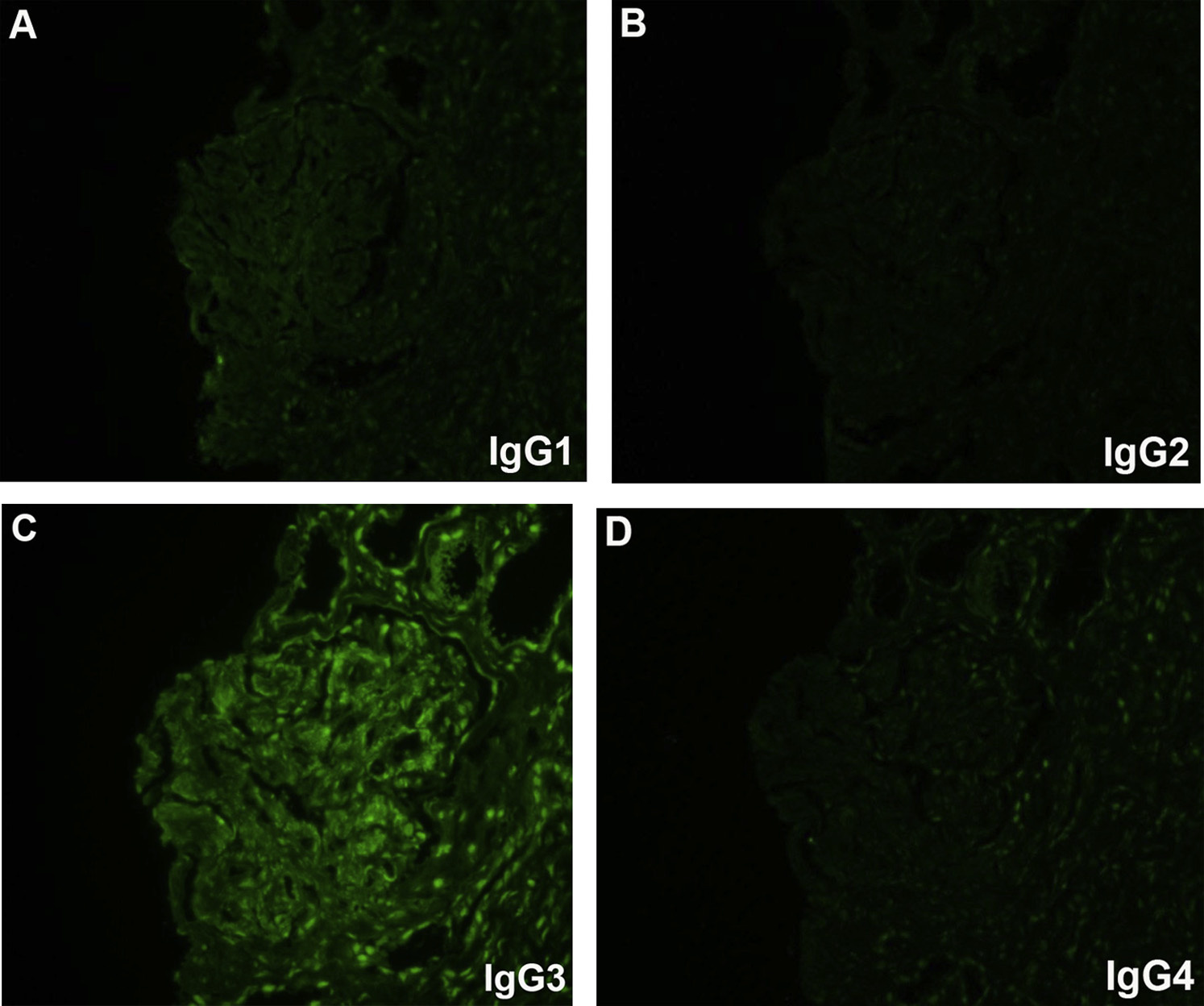
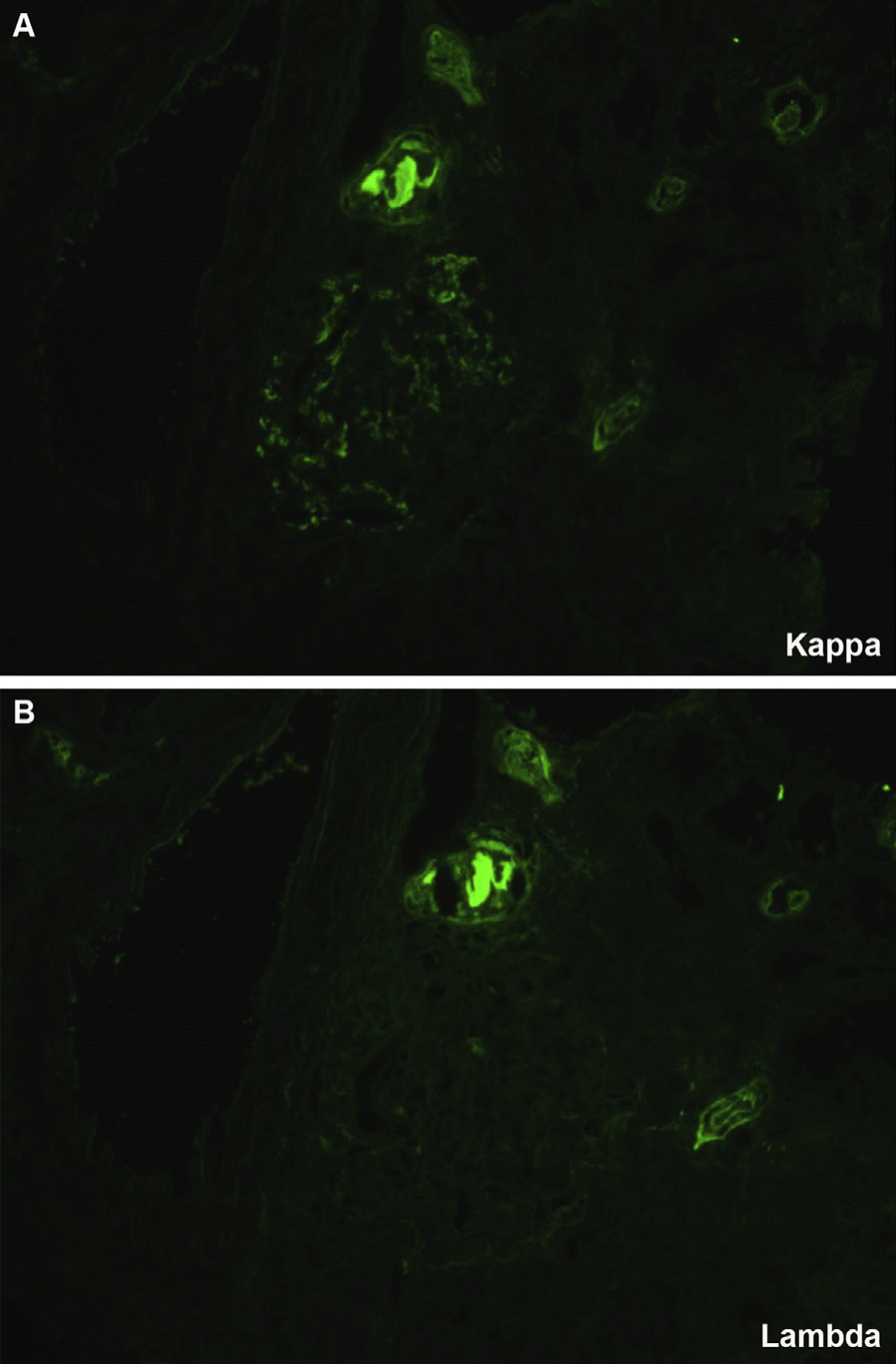
Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) is a condition where monoclonal immunoglobulins are deposited in the glomerulus. Patients are typically older than 50 years at presentation, but there is a wide age range (20-81 y). Presenting features include nephrotic-range proteinuria in about half, hematuria in three-fourths, and reduced glomerular filtration rate in two-thirds.
Only a third have detectable circulating monoclonal protein at presentation, and multiple myeloma is rare, even during follow-up. Approximately a third of patients with PGMID completely or partially improve, a third have persistent chronic kidney disease (CKD), and ~20% progress to end-stage kidney disease.
Markers of poor prognosis are more elevated serum creatinine at presentation, and the degree of glomerulosclerosis and interstitial fibrosis. PGNMID may recur in the kidney transplant, despite lack of detectable circulating monoclonal protein.
Light microscopy: Predominantly membranoproliferative lesions, often with glomerular basement membrane double contours, and occasionally only with mesangial proliferation. A membranous pattern has rarely been observed.
Immunofluorescence microscopy: Deposits most often stain mesangial areas and glomerular capillary walls in an irregular, coarsely granular pattern.
Monoclonal immunoglobulin G3 (IgG3), most typically with κ light chain restriction, is most common, with IgG1κ subclass next. There are associated C3 deposits, and C1q may also be present. Rare cases with membranous pattern by light microscopy show corresponding finely granular capillary wall staining.
No extraglomerular IgG deposits are present.
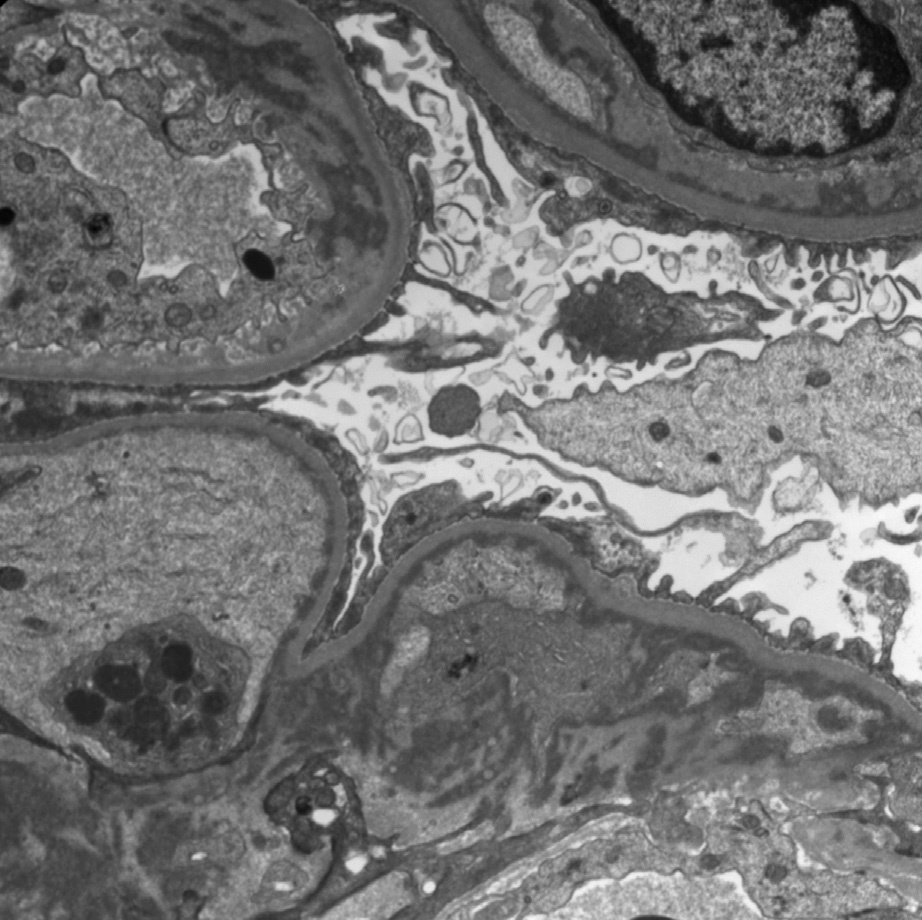
Electron microscopy: There are predominantly mesangial and subendothelial deposits, often indistinguishable from deposits of immune complexes in glomerulopathies unassociated with lymphoplasmacytic disorders. Rare membranous pattern cases show subepithelial deposits. Deposits are usually not organized, and there are no tubular basement membrane deposits.
PGNMID is caused by monoclonal immunoglobulin deposition localized to glomeruli. The culprit is most often IgG3κ, although this is the rarest of the 4 IgG isotypes. IgG3 is nephritogenic and fixes complement. Glomerular capillary wall localization is favored by its high molecular weight and anionic charge. No mutations of the constant regions of IgG3, or other IgG, have been detected, unlike in heavy chain deposition disease (HCDD).
PGNMID is differentiated from membranoproliferative glomerulonephritis resulting from immune complexes (eg, lupus nephritis, chronic infections) because there is polyclonal staining of deposits (ie, both κ and λ) in the latter. HCDD shows only heavy chain staining, contrasting both light and heavy chain monoclonal staining in PGNMID, and most often with concomitant tubular staining and occasional vascular staining. Light and HCDD (LHCDD) most often shows tubular and occasional vascular deposits, contrasting the absence of extraglomerular deposits in PGNMID. Further, the deposits in PGNMID are indistinguishable from usual immune complex deposits by electron microscopy, contrasting powdery amorphous granular deposits in LHCDD.