Microscopy Images
Congenital nephrotic syndrome of Finnish type presents as nephrotic syndrome. Patients typically present at birth or within the first 3 months of life, and very rarely beyond 1 year of age. Microscopic hematuria is often present. Patients may die of complications of massive nephrotic syndrome unless treated, sometimes with pharmacologic or surgical nephrectomy to stop massive protein loss, in addition to renal replacement therapy and transplantation.
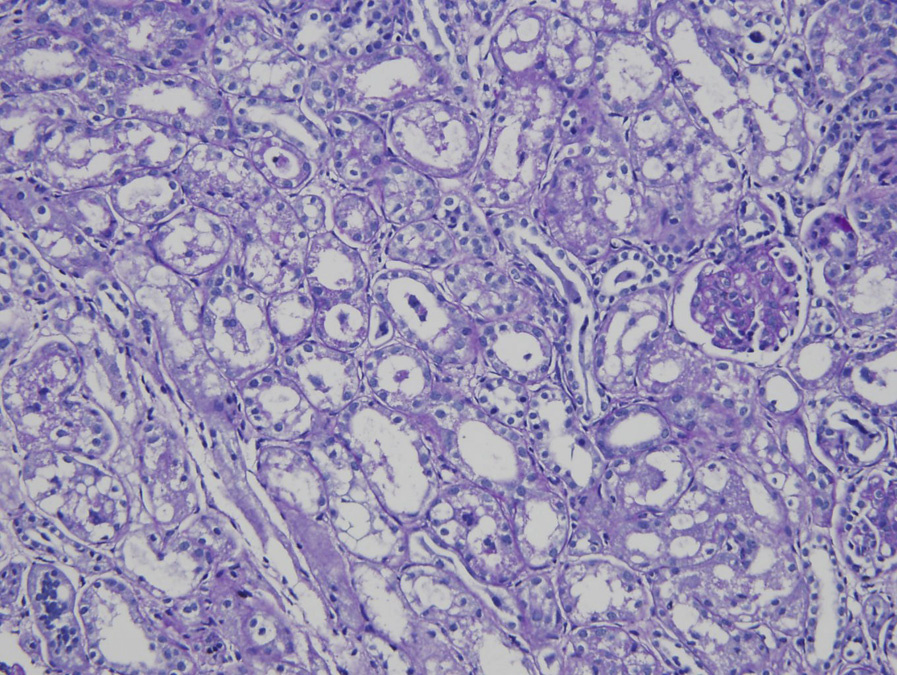
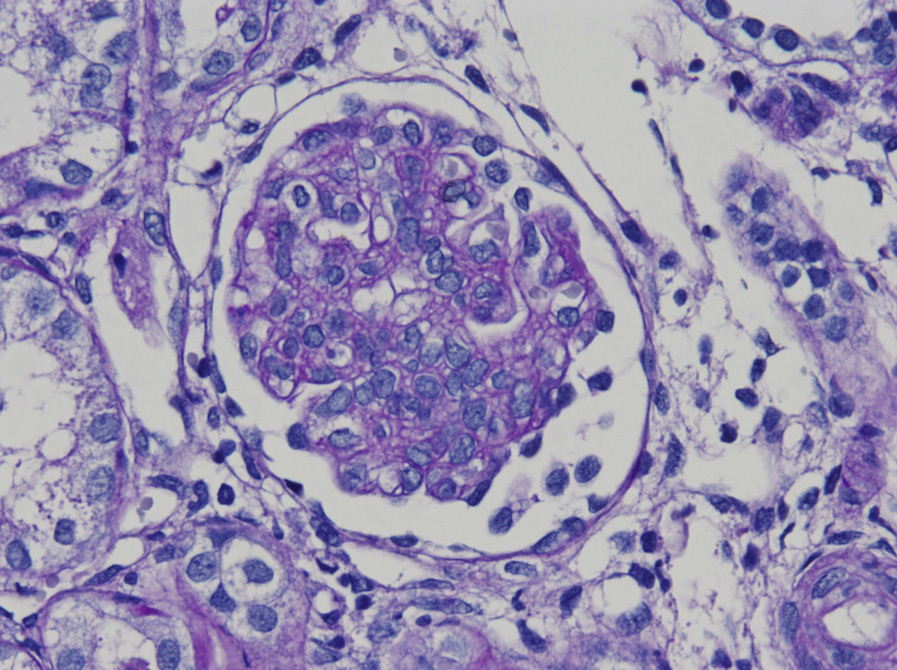
Light microscopy: Glomeruli may be unremarkable by light microscopy or show variable mesangial proliferation and/or endocapillary hypercellularity, with occasional crescents but without necrosis or glomerular basement membrane breaks. Nonspecific glomerulosclerosis develops late in the course. Proximal tubules are dilated.
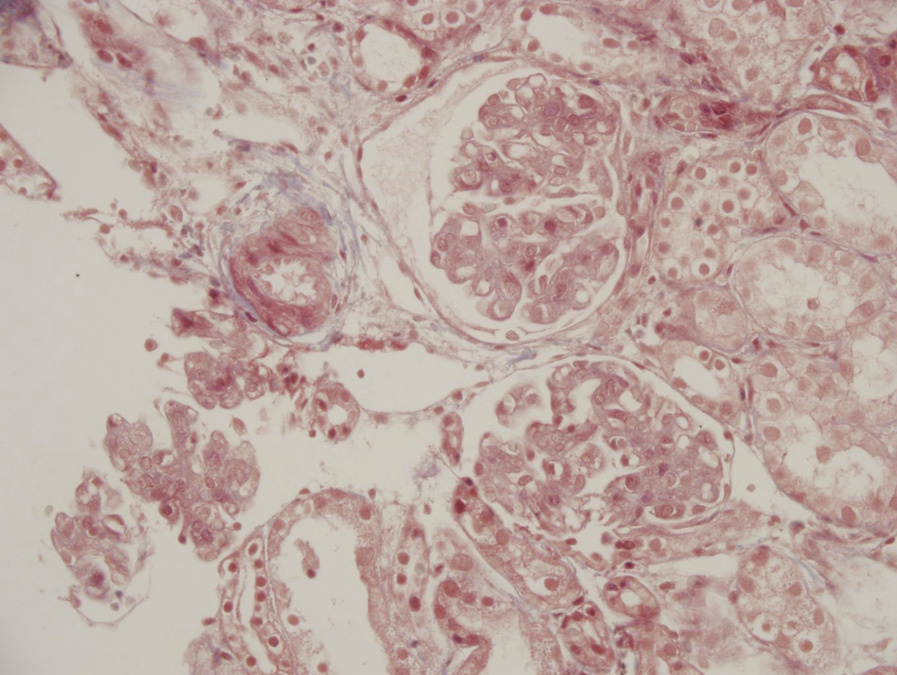
Immunofluorescence microscopy: No specific staining.
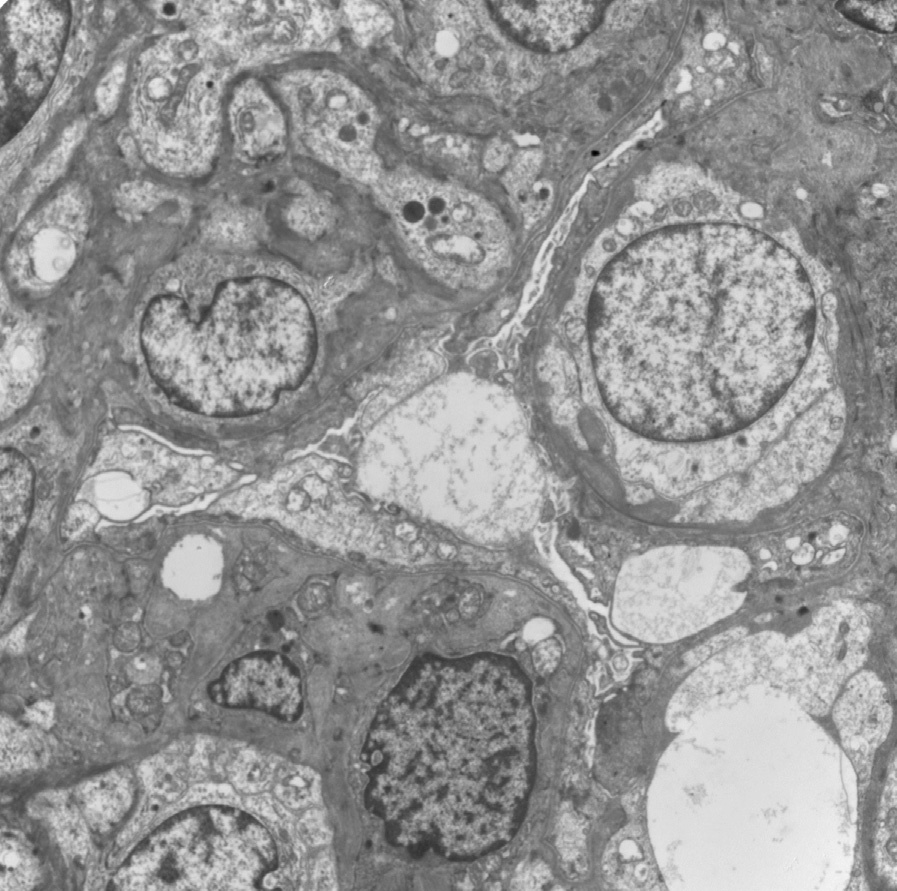
Electron microscopy: Complete foot process effacement.
Congenital nephrotic syndrome of Finnish type is an autosomal recessive disease due to mutations in nephrin (NPHS1), a key component of the podocyte slit diaphragm.
Morphologic findings are not specific, and genetic analysis is needed to confirm the diagnosis. The proliferative lesions may suggest immune complex disease, but immunofluorescence and electron microscopy studies show absence of immune complex deposits in congenital nephrotic syndrome of Finnish type.
Diffuse mesangial sclerosis may also cause nephrotic syndrome at this age, but typically demonstrates a distinct increase in mesangial collagen.