Microscopy Images
Lupus nephritis (LN) is defined as glomerular immune complex disease that occurs in patients who meet American College of Rheumatology for diagnosis of systemic lupus erythematosus (SLE). SLE is a systemic autoimmune disease, most commonly involving the skin, kidneys, joints, heart, and serosal surfaces. Women are affected more than men (9:1), and SLE is more common in African Americans.
Onset usually is from teenage years to the third decade of life, but SLE may manifest at any age.
Kidney involvement is a major cause of morbidity and the most common cause of death in SLE patients.
The varying glomerular immune complex patterns are diagnosed according to the ISN/RPS classification.
These include predominantly mesangial deposits (classes I, II), subendothelial deposits with endocapillary hypercellularity or prominent duplication of glomerular basement membranes (GBMs), often with necrotizing and crescentic lesions (classes III and IV, focal and diffuse LN), or membranous forms (class V). Focal LN (class III) is present in 20%-35% of biopsied SLE patients, with less than half of glomeruli with lesions as specified below. Diffuse LN (class IV) comprises the same lesions as in focal LN, but involves half or more of glomeruli, and is diagnosed in ~30%-60% of biopsied SLE patients. Patients with focal LN may have only mild proteinuria and microscopic hematuria or have acute kidney injury, more proteinuria, and red blood cell casts. Patients with diffuse LN typically have more proteinuria, hematuria, with associated hypertension and decreased kidney function, and worse prognosis, which if not treated commonly results in end-stage kidney disease.
However, there can be substantial clinical overlap in the presentation of patients with focal and diffuse LN. LN may precede other findings of SLE, or develop at any time in the disease course, and manifests initially with any class. Transformations between classes—spontaneously or in response to therapy—are common.
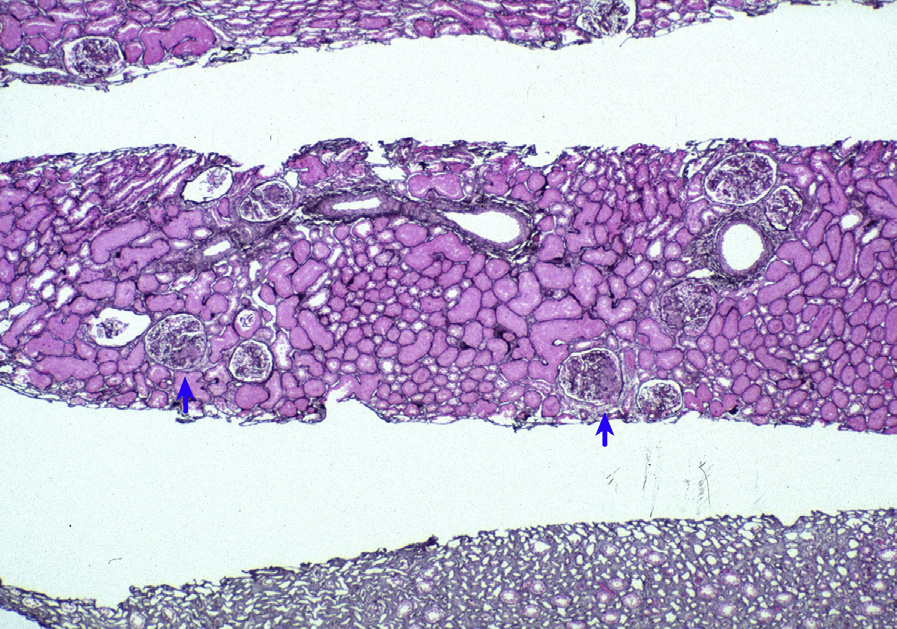
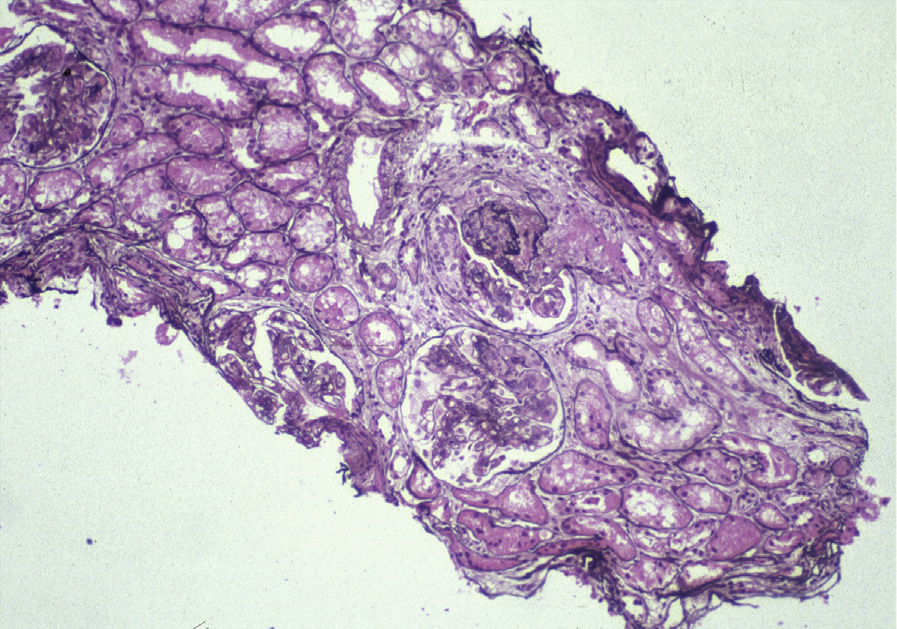
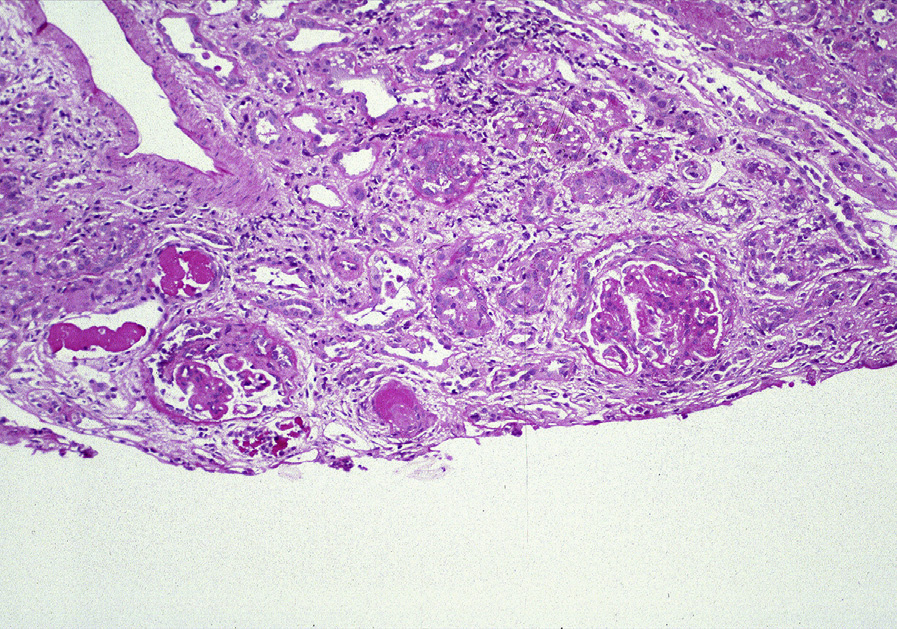
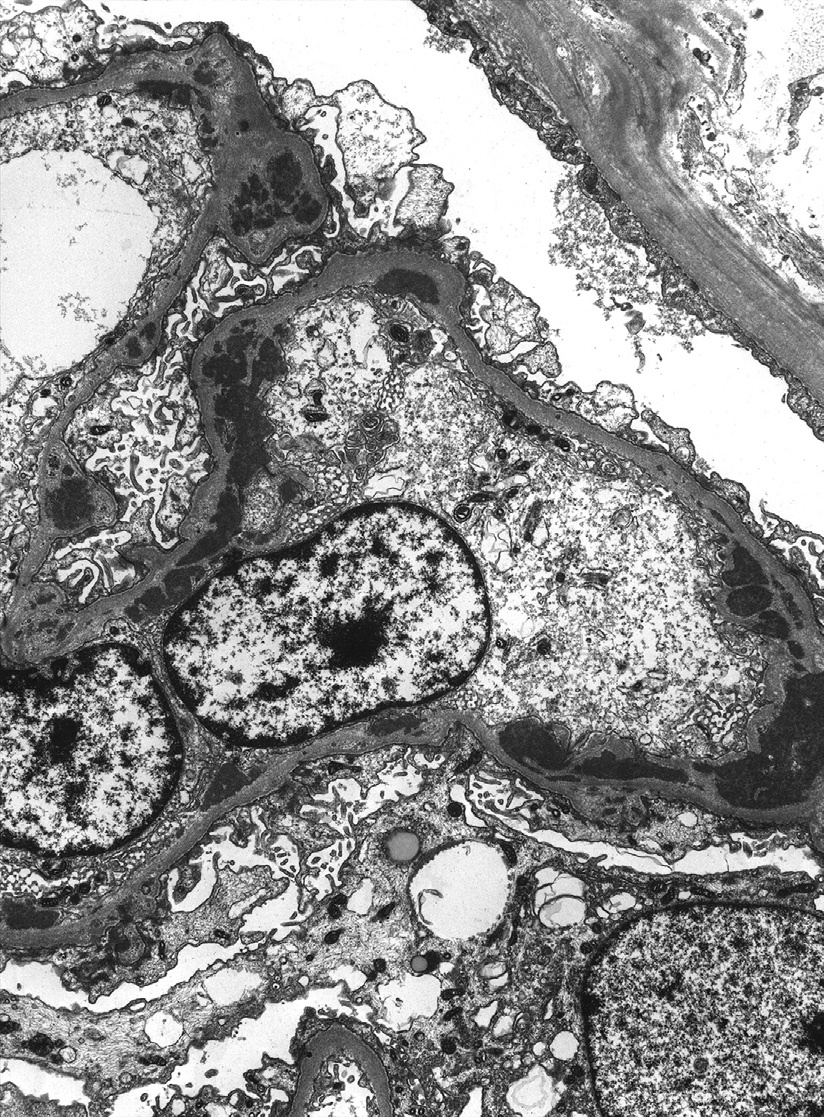
Light microscopy: Focal and diffuse LN are characterized by segmental or global endocapillary and mesangial hypercellularity, often with infiltration by inflammatory cells, with or without fibrinoid necrosis or crescents. Glomeruli often exhibit a membranoproliferative pattern of injury. Extensive subendothelial deposits result in a wire loop appearance, associated with double contours of GBMs.
Large subendothelial deposits bulging into the lumen, so-called hyaline thrombi, may be present. Segmental necrotizing lesions with fewer deposits than expected, resembling the lesions of pauci-immune glomerulonephritis (GN), may be present, especially in focal LN. In addition, chronic lesions of fibrocellular or fibrous crescents and segmental glomerular scars with broad-based adhesions may result from organization of the above active injuries. Focal LN is diagnosed when less than half of glomeruli are involved with the above active or chronic lesions; diffuse LN, when at least half of glomeruli are involved. Activity (endocapillary hypercellularity, necrosis, presence of wire loops and/or hyaline thrombi, cellular or fibrocellular crescents, and active interstitial inflammation) and chronicity (segmental or global glomerulosclerosis due to LN, fibrocellular or fibrous crescents, and tubulointerstitial fibrosis) should be specified. In addition, class VI LN is diagnosed when >90% of glomeruli show sclerosis with no residual activity.
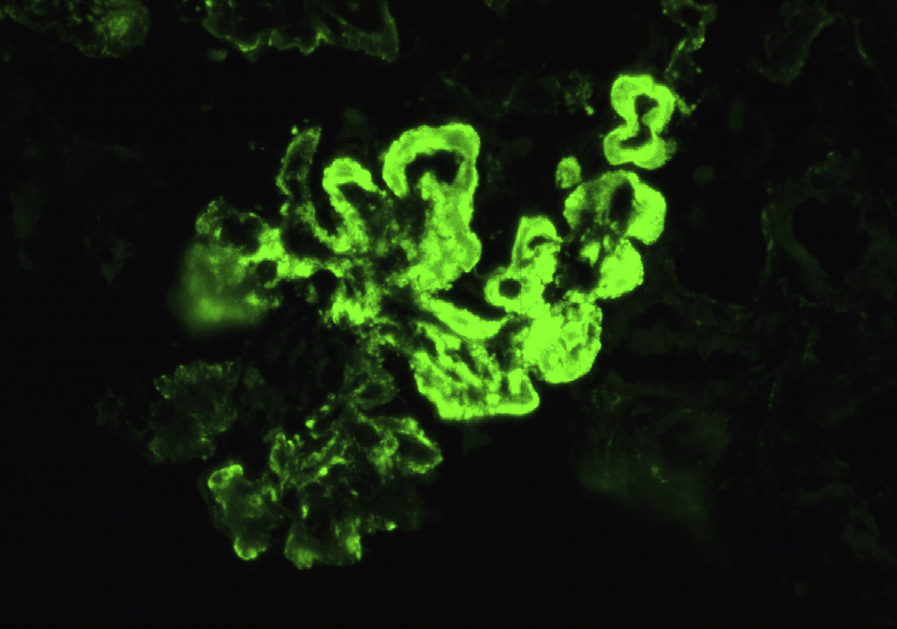
Immunofluorescence microscopy: There is often “full-house” staining with polyclonal IgG, IgA, IgM, C3, and C1q, with IgG dominance or codominance; staining is diffuse and global in the mesangium with varying glomerular capillary loop extension, with chunky, sausage-like, or wire loop lesions corresponding to the subendothelial deposits. Staining may be restricted to IgG and complement. Nuclear staining for IgG, reflecting positive ANA, may be present.
Granular immune complex deposits more commonly involve peritubular capillaries than in other classes of LN, particularly with staining for C1q. Granular tubular basement membrane deposits, either focal or diffuse, occur commonly, with variable lymphoplasmacytic infiltrates. Bland vascular deposits in arterioles and arteries are common.
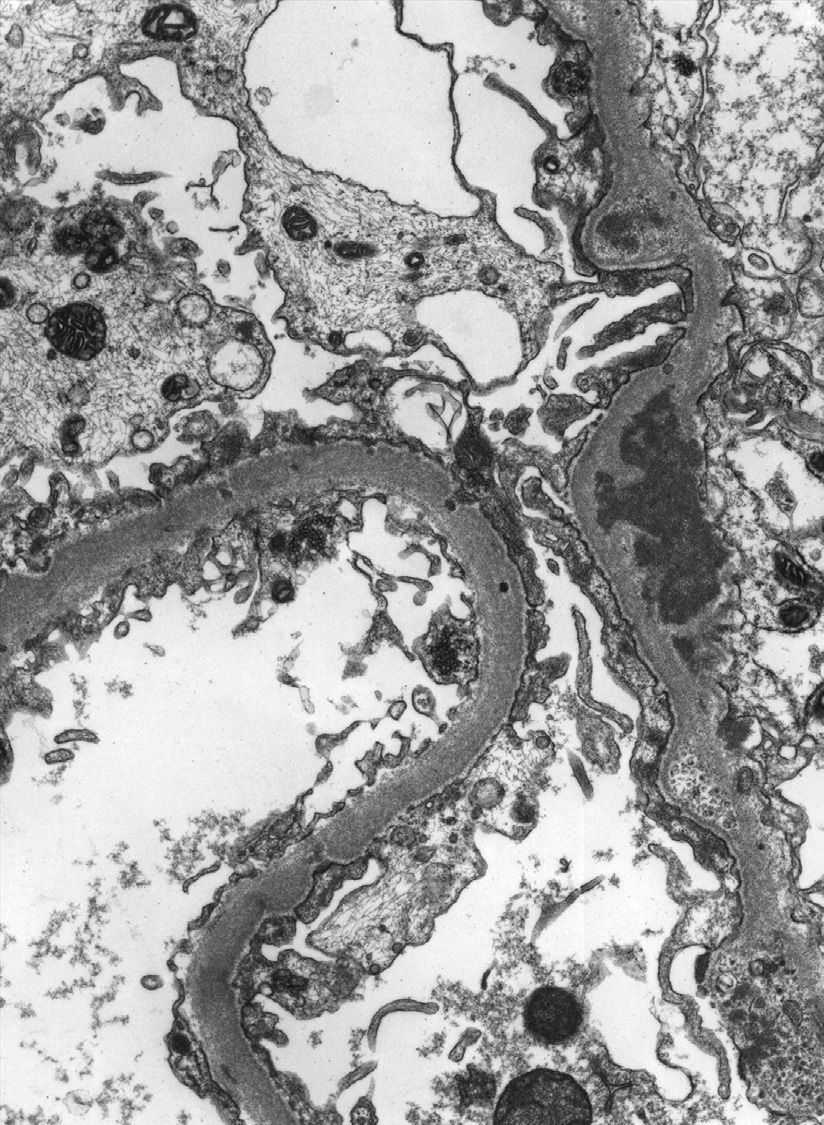
Electron microscopy: There is endocapillary and mesangial hypercellularity due to mesangial and varying subendothelial deposits, with tubuloreticular aggregates (a response to high interferon levels) in endothelial cell cytoplasm. Scattered subepithelial deposits, involving less than half the glomeruli and less than half the loops, may be present. If most glomeruli and loops show subepithelial deposits, additional membranous LN (class V) should be diagnosed. The deposits may exhibit substructural organization, with appearances as fibrils and/or whorled “fingerprints.”
There are genetic predispositions to SLE. Inadequate clearance of nuclei from cells undergoing apoptosis (triggered by various environmental insults, including ultraviolet radiation) leads to excess nuclear antigens which, combined with additional abnormalities in B and T lymphocytes, ultimately results in autoantibody formation. The activation of Toll-like receptors and dendritic cells and high interferon a levels are additional elements postulated to perpetuate this process. Autoantibodies develop against a variety of nuclear materials, including DNA, RNA, and various histone and nonhistone proteins. Autoantibodies may bind to endogenous intrinsic antigens, or exogenous antigens “planted” from the circulation, with in situ immune complex formation. The site of immune complex deposition determines the injury response: mesangial deposits lead to mesangial hypercellularity, subendothelial deposits cause the endocapillary hypercellularity in focal and diffuse LN, and subepithelial deposits cause a response of podocytes in membranous LN. SLE with LN may be induced by some drugs, notably propylthiouracil, hydralazine, procainamide, and isoniazid. Full-house immune complex GN may occur in some HIV-infected patients without clinical evidence of SLE.
Renal deposits may also occur in extraglomerular locations. Vascular lesions are varied, including bland, uncomplicated vascular deposits, and thrombotic microangiopathy, the latter typically in combination with focal or diffuse LN.
Thrombotic microangiopathy due to circulating antibodies to phospholipids or cardiolipin with or without immune complex deposition may occur. Necrotizing lupus vasculitis is very rare, with fibrinoid necrosis, transmural leukocytes, and variable immune deposits. In some patients, very segmental necrotizing glomerular lesions with less hypercellularity and proliferation and deposits than expected suggest possible additional pauci-immune ANCA-related mechanisms.
Focal or diffuse immune complex GN, often with proliferative lesions, may be due to infection, lupuslike condition, C3 GN, membranoproliferative GN, cryoglobulinemia, proliferative GN with monoclonal deposits, or C1q nephropathy, among others, each with disease-specific findings, but typically lacking full-house staining with IgG dominance or codominance and tubuloreticular aggregates. Full-house staining of immune complex deposits can also be seen in shunt nephritis. HIV infection can cause immune complex GN with features mimicking LN.