Microscopy Images
Heavy chain deposition disease (HCDD) is the rarest of the monoclonal immunoglobulin deposition diseases (MIDD), and is diagnosed by immunofluorescence or immunohistochemistry. The reported age range of HCDD is from 35-79 years with men and women equally affected, and one-third of patients demonstrating hypocomplementemia. Patients present with proteinuria, often in the nephrotic range, hematuria, hypertension, and reduced glomerular filtration rate. The prognosis is dependent on the underlying dysproteinemic disorder. Most patients have progressive glomerular filtration rate loss. Recurrence of MIDD has been reported in the transplant.
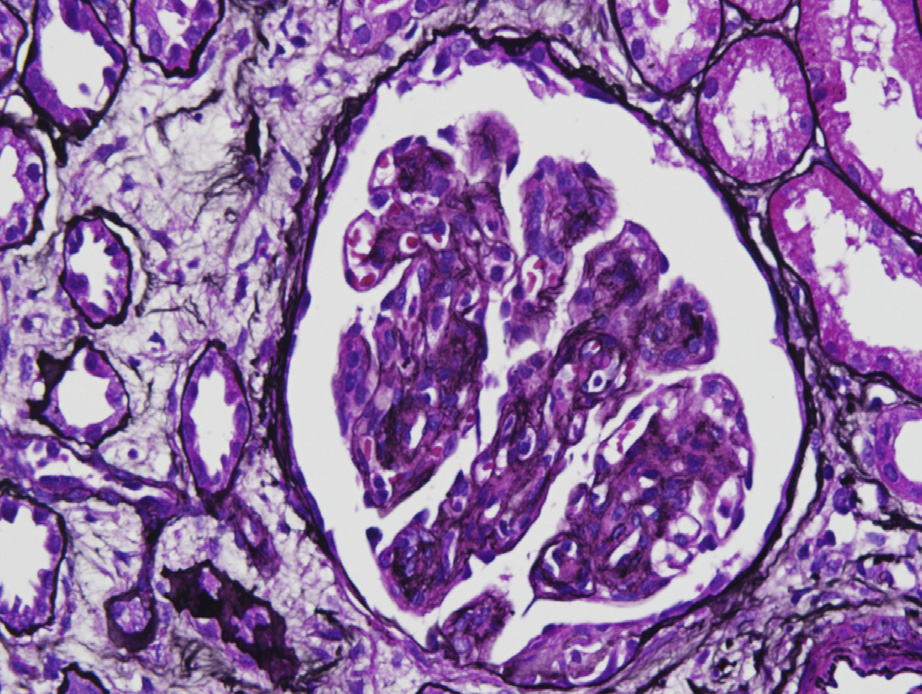
Light microscopy: HCDD typically shows nodular sclerosis, with variable crescents in some cases.
Nearly all cases demonstrate glomerular deposits, with most cases also showing deposits along tubular and arterial wall basement membranes. Glomerular basement membrane double contours may occur.
Tubulointerstitial fibrosis and atrophy are proportional to glomerular injury. Congo Red stain is negative.
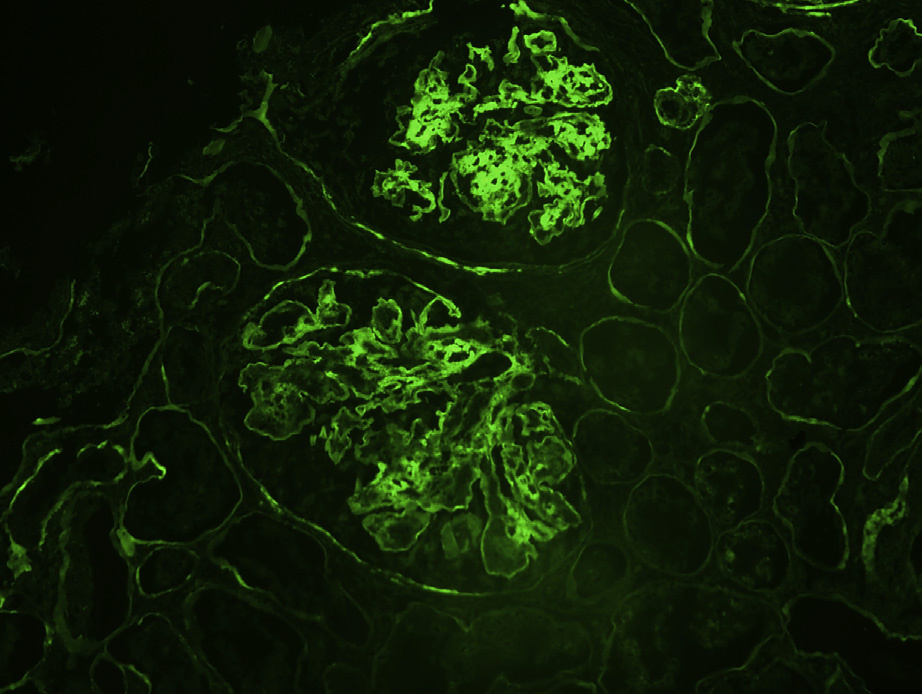
Immunofluorescence microscopy: The key finding is monoclonal heavy chain staining, most frequently with g heavy chain, in a pseudolinear or occasional granular fashion along glomerular, and often tubular, and occasional arterial wall basement membranes. A minority of cases also show concomitant complement C3 deposits.
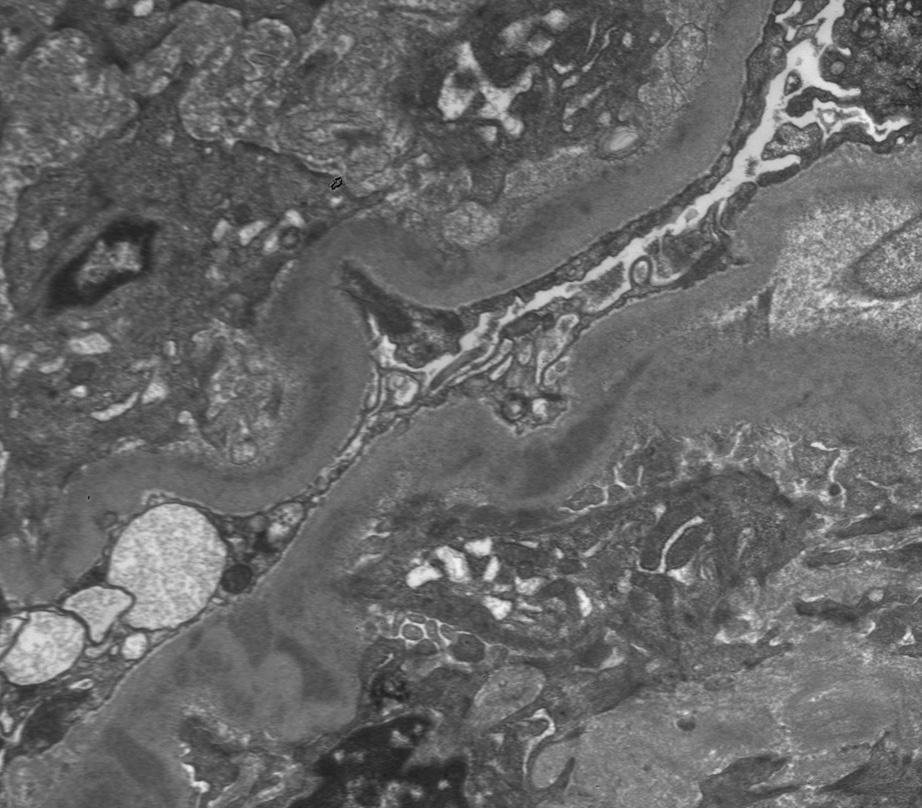
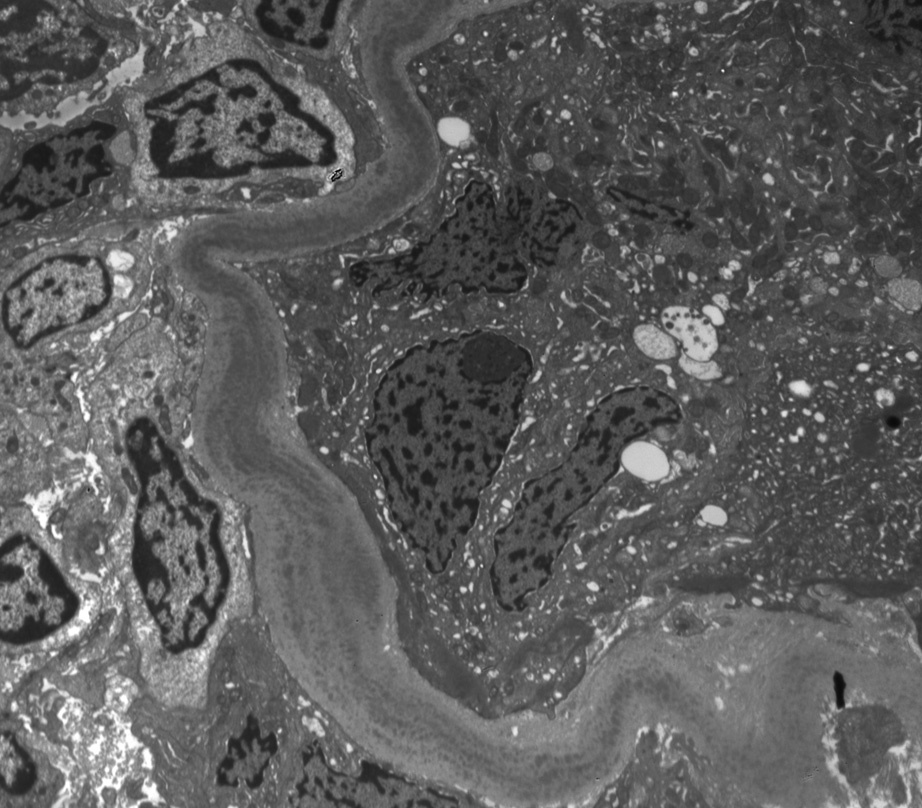
Electron microscopy: Deposits are present along glomerular, tubular, and arterial wall basement membranes, corresponding to deposits seen on immunofluorescence. These may be finely granular, punctate, powdery, or ground pepper–like, with occasional substructure observed in some cases. Occasional cellular interposition results in double-contour appearance of the glomerular basement membranes.
HCDD is due to deposition of monoclonal heavy chain in mesangial areas and along glomerular, tubular, and/or arterial basement membranes. The monoclonal protein is due to an underlying plasma cell dyscrasia. In some cases, it has been shown that the heavy chain deposits represent a truncated heavy chain with deletion of CH1, preventing binding of the heavy chain to heavy chain–binding protein in the endoplasmic reticulum and impairing assembly with light chains. The truncated abnormal heavy chains have a predisposition for tissue deposition, and are therefore usually not found in the urine. Monoclonal protein is most often detected in the serum, without detectable isolated monoclonal heavy chain in the urine.
HCDD may be distinguished from other causes of nodular sclerosis and mesangial expansion by specific heavy chain staining in glomerular and tubular basement membranes, negative Congo Red stain, and characteristic punctate, amorphous, or ground pepper–like appearance of deposits on electron microscopy. Fibrillary glomerulonephritis is Congo Red negative, and usually manifests as deposits of polyclonal immunoglobulin G; diabetic nephropathy shows no deposits, and other types of MIDD show staining restricted either to one light chain subclass only or to one heavy and light chain subclass only. Other causes of a membranoproliferative glomerulonephritis pattern show disease-specific immunofluorescence and electron microscopic appearances.