Microscopy Images
Light and heavy chain deposition disease (LHCDD) occurs primarily in adults with an average age of 55-60 years, presenting with proteinuria (often in the nephrotic range), hematuria, hypertension, and reduced glomerular filtration rate. LHCDD is the second most common form of the non-AL amyloid monoclonal immunoglobulin deposition diseases (MIDDs), accounting for about 10% of these patients.
The specific type of MIDD is diagnosed by immunofluorescence or immunohistochemistry. The prognosis is dependent on the underlying cause of dysproteinemia. There is limited experience with transplantation, but recurrence of MIDD has been reported in a transplant.
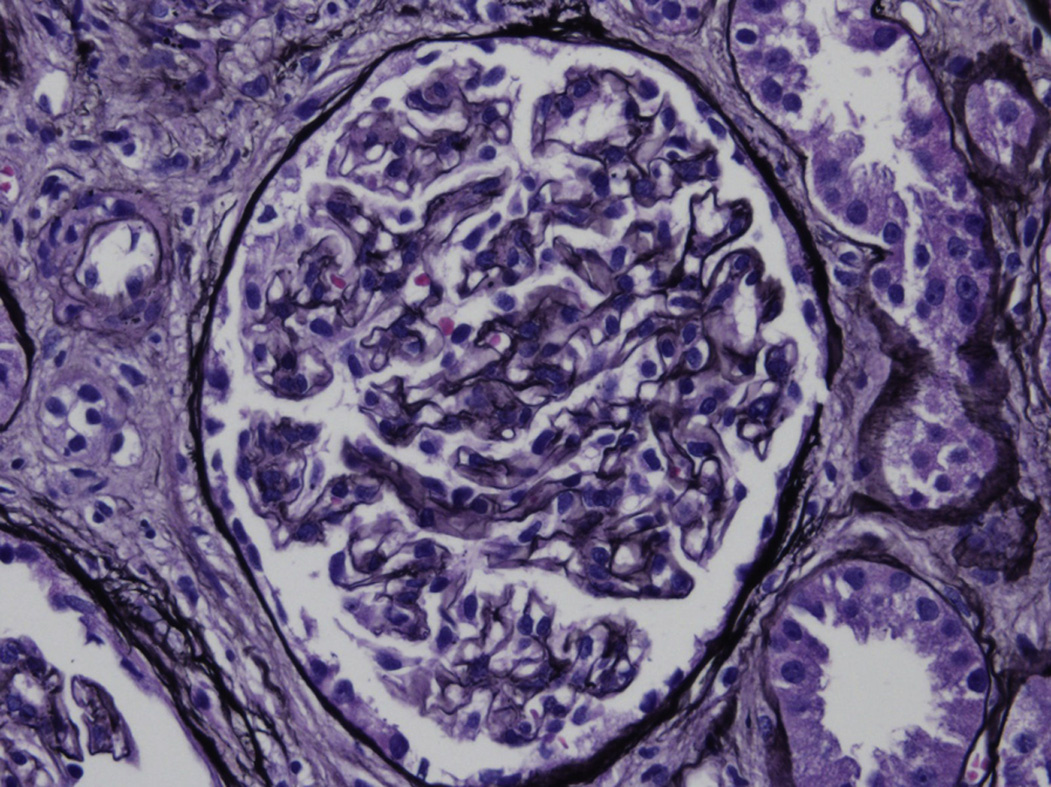
Light microscopy: The appearance of LHCDD is variable, ranging from unremarkable glomeruli to mesangial proliferative, membranoproliferative, or nodular mesangial appearances. Crescents have been rarely reported, and Congo Red stain is negative.
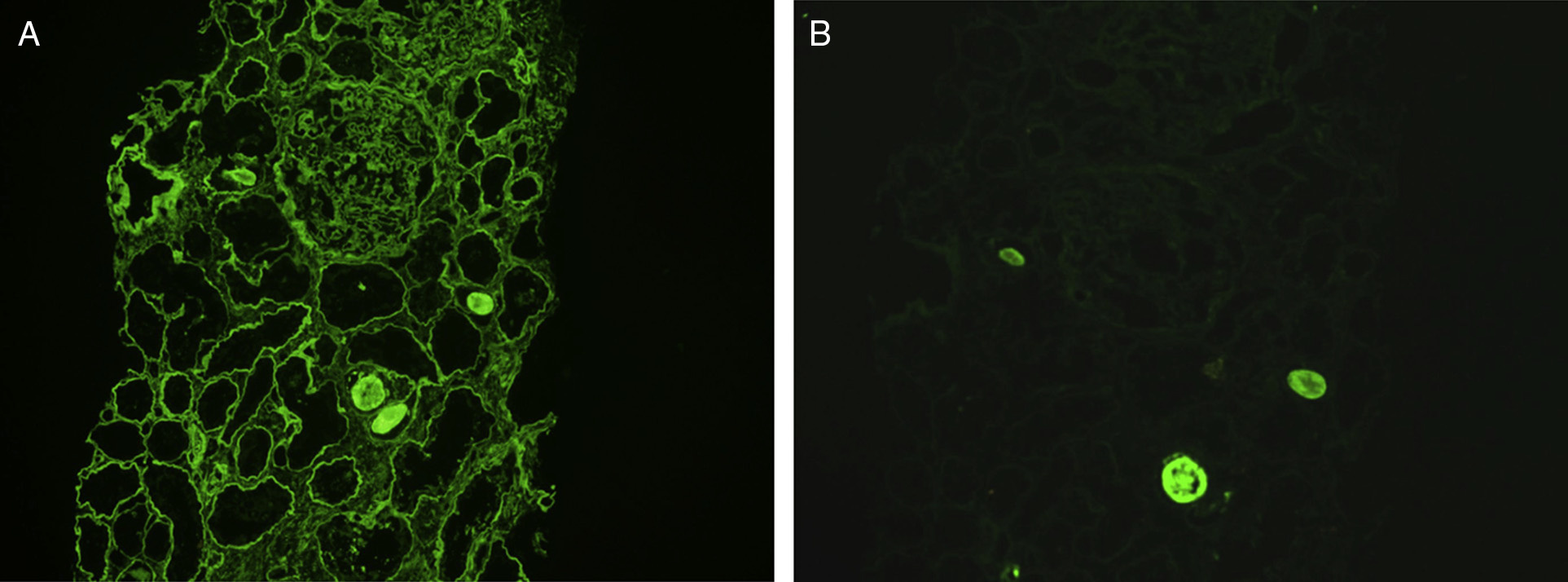
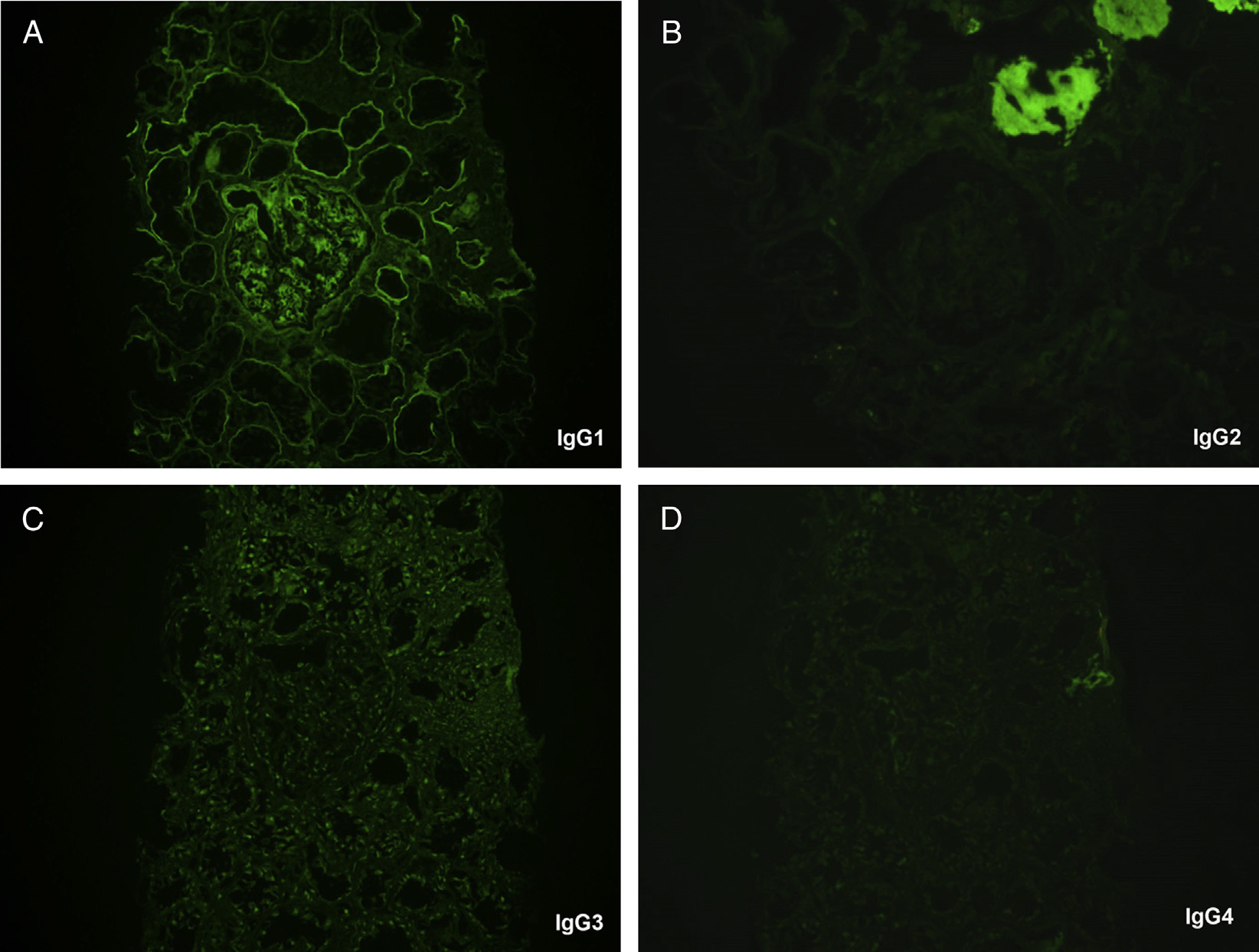
Immunofluorescence microscopy: Monoclonal light chain stain with either κ or λ light chain, and most frequently g heavy chain. Staining is linear, or less commonly granular, along glomerular and tubular basement membranes, with additional staining in the mesangium. Immunoglobulin G (IgG) subtype staining confirms clonality of the heavy chain component in cases with g heavy chain.
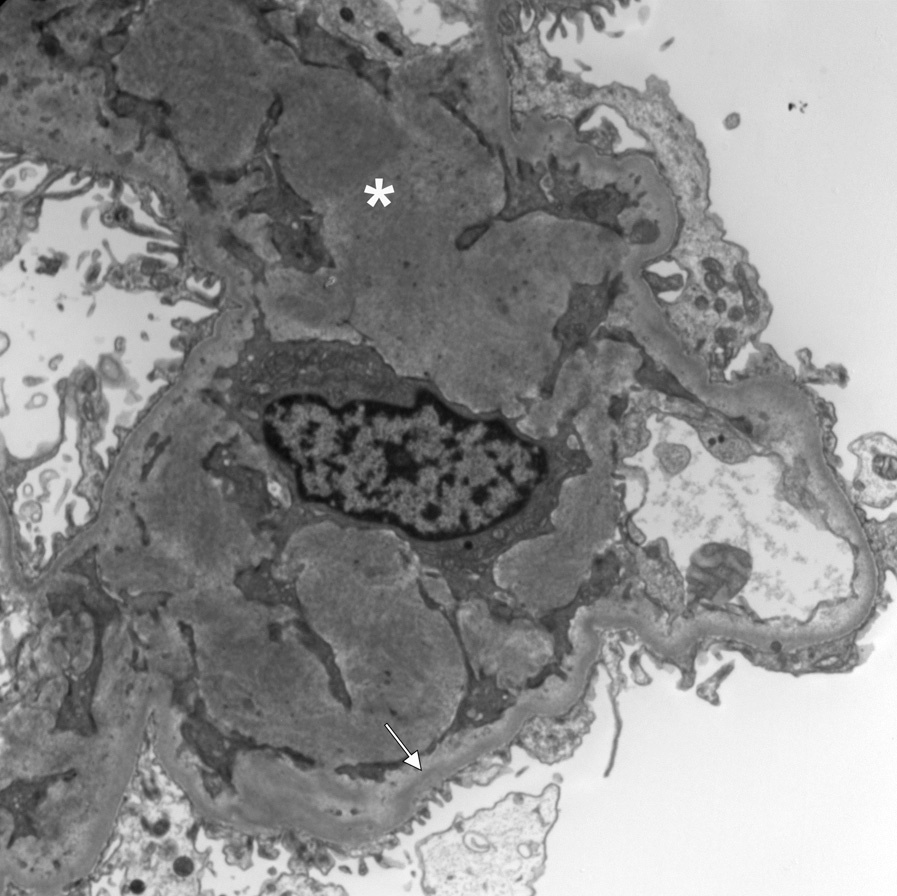
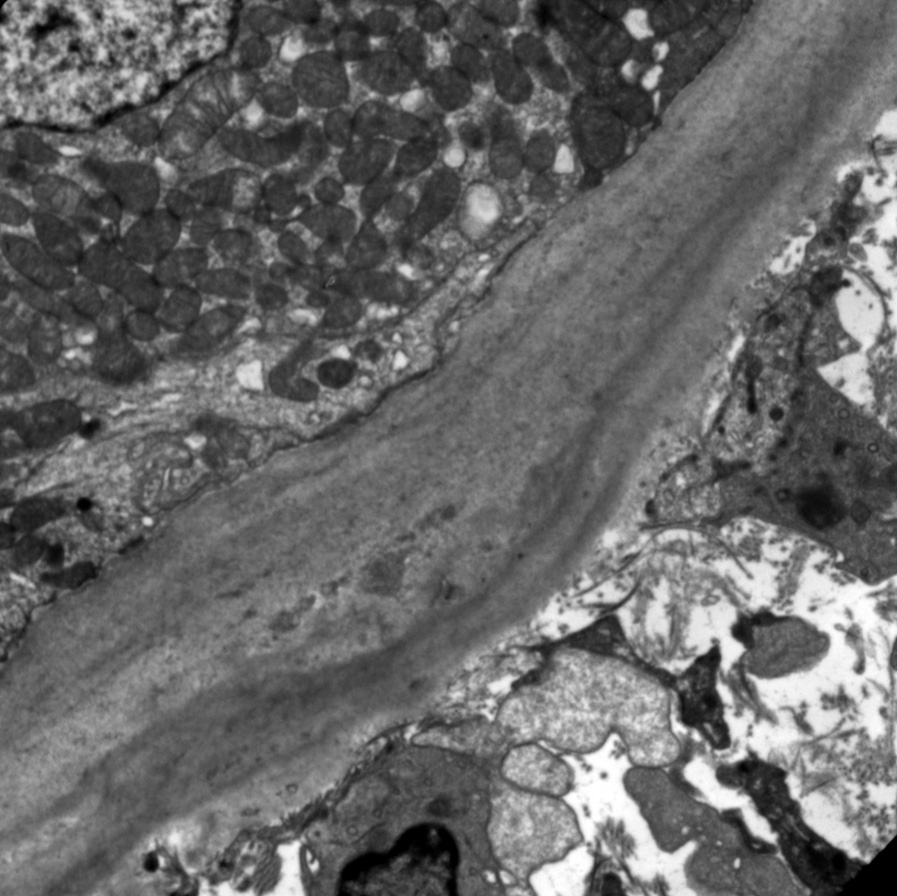
Electron microscopy: Variable deposits correspond to the light microscopic pattern, with mesangial and inner glomerular and tubular basement membrane deposits of extracellular electron dense material.
There is sometimes a fibrillar substructure, more commonly with punctate, powdery, ground pepper–like deposits.
LHCDD is due to deposition of monoclonal light and heavy chain in mesangial areas and along glomerular and tubular basement membranes. The monoclonal protein is due to an underlying plasma cell dyscrasia. About half of patients have multiple myeloma. In a limited number of cases in which this has been studied, the abnormal heavy chains have deletions in the heavy chain portion, CH1 or CH2, which result in early secretion of these immature forms into the circulation and deposition in the kidney. These CH1 domains usually bind to heavy chain binding protein to allow IgG assembly in the endoplasmic reticulum.
LHCDD may be distinguished from other causes of nodular sclerosis and mesangial expansion by specific light and heavy chain staining in glomeruli and tubular basement membranes, negative Congo Red stain, and characteristic punctate, amorphous, ground pepper–like appearance of deposits on electron microscopy. Fibrillary glomerulonephritis is Congo Red negative, and usually manifests as deposits of polyclonal IgG. Diabetic nephropathy shows no deposits, and other types of MIDD show staining restricted either to one light chain subclass or to one heavy chain subclass. Other causes of a membranoproliferative glomerulonephritis pattern show disease-specific immunofluorescence and electron microscopic appearances.