Microscopy Images
IgG4-related disease is an immune-mediated systemic condition characterized by presence of discrete mass lesions or diffuse infiltrative lesions in organs, composed of lymphoproliferative infiltrates including frequent IgG4-producing plasma cells and variable storiform fibrosis. Serum levels of IgG4 are increased (>135 mg/dL) in ~2/3 of patients. It is more common in middle-aged and older men. In 60-90% of patients, multiple organs/sites are involved. The most common sites include pancreas, biliary tract, salivary or lacrimal glands, orbits, retroperitoneum, and kidneys. Lymphadenopathy is common. About 40% of patients have history of asthma or allergy and over 1/3 have peripheral eosinophilia. Kidney involvement (IgG4-related kidney disease) includes tubulointerstitial nephritis (TIN), membranous nephropathy (MN), and pyelitis. The disease is indolent and may be asymptomatic, but patients may also present with proteinuria, hematuria, decreased kidney function, and hypocomplementemia (reduced C3 and C4 in ~50% of patients). Nephrotic-range proteinuria often occurs in patients with MN. Imaging may show multiple, often bilateral, tumoral lesions in the kidneys, and thickening of pelvis wall if pyelitis is present. Favorable response to glucocorticoids, especially if started prior to emergence of extensive fibrosis, is characteristic.
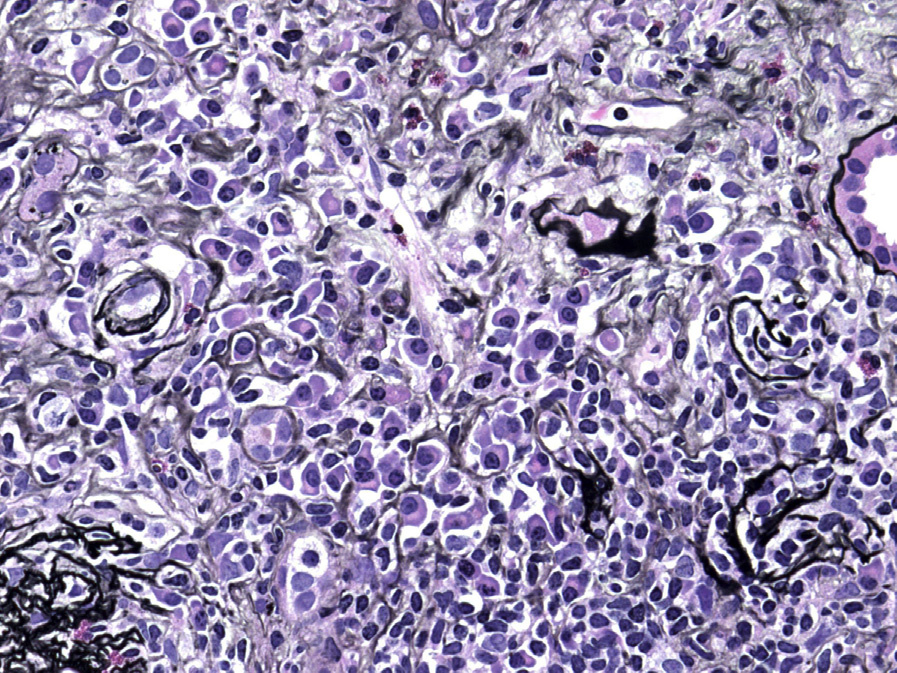
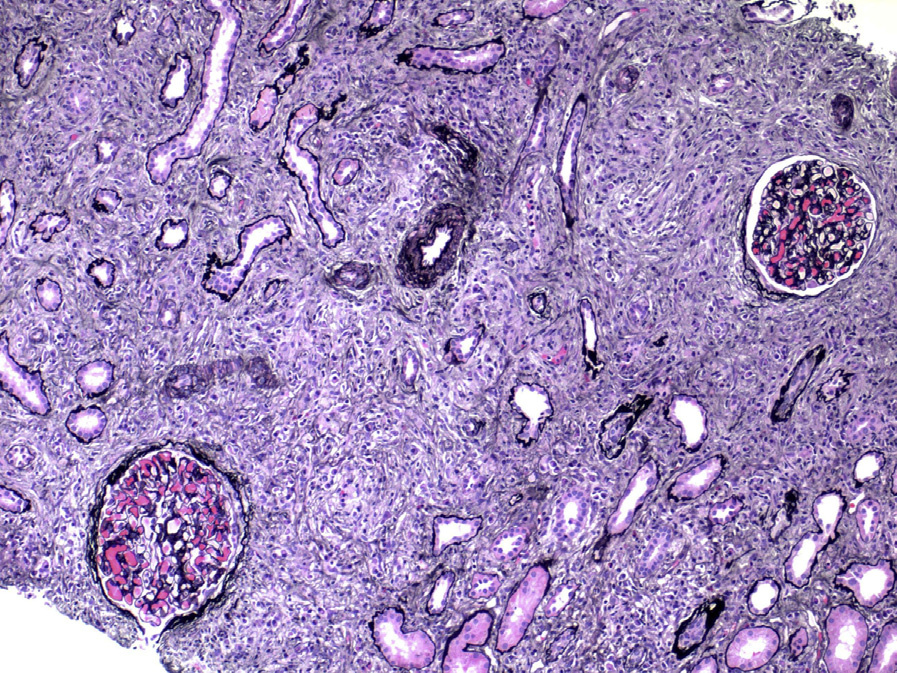
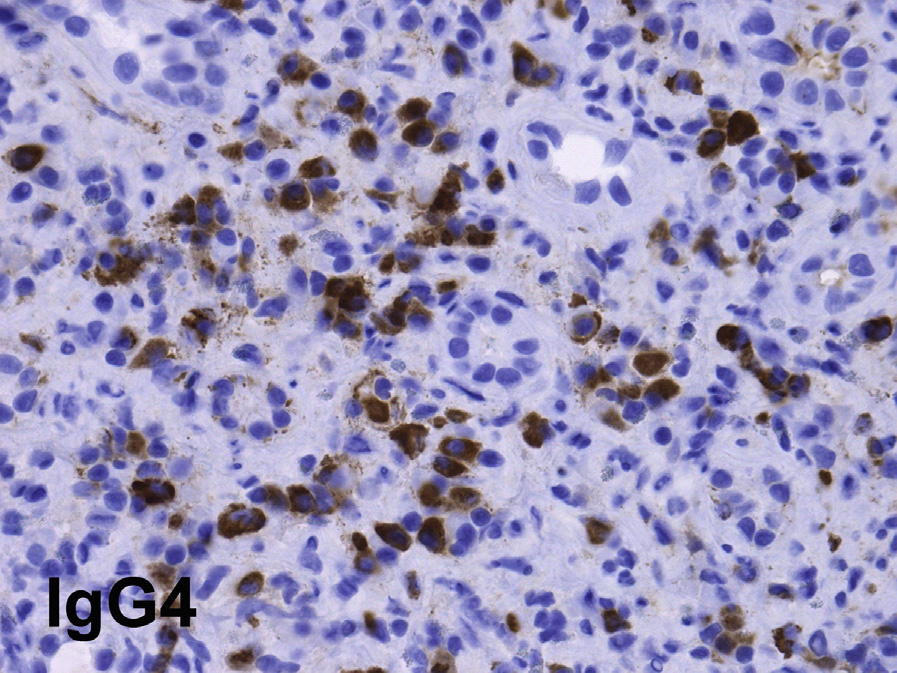
Light microscopy: TIN is the most common finding in IgG4-related kidney disease. Dense lymphoplasmacytic infiltrates with a predominance of IgG4-positive plasma cells (>10 IgG4-positive plasma cells per high power field, and/or >40% IgG4/IgG-positive plasma cells in the most affected areas), usually accompanied by interstitial fibrosis with a “storiform” pattern (swirling fibrosis with fibroblasts arranged like spokes of a cartwheel) is the most typical injury pattern. Bird’s eye fibrosis, characterized by nests of inflammatory cells surrounded by irregular collagen fibers, best seen with periodic acid–Schiff and Jones silver stains, is unique. Frequent eosinophils may be present. Tubular atrophy and dropout are common in fibrotic regions. The margin between affected and unaffected areas is often well demarcated. Extension of the inflammation into the renal capsule is typical. Obliterative phlebitis is rarely seen in the kidney. The most common concomitant glomerular lesion is MN, seen in 7-10% of cases.
IgA nephropathy (Henoch-Schönlein purpura), endocapillary proliferative glomerulonephritis, membranoproliferative glomerulonephritis, and mesangial proliferative glomerulonephritis can also be seen in IgG4-related kidney disease.
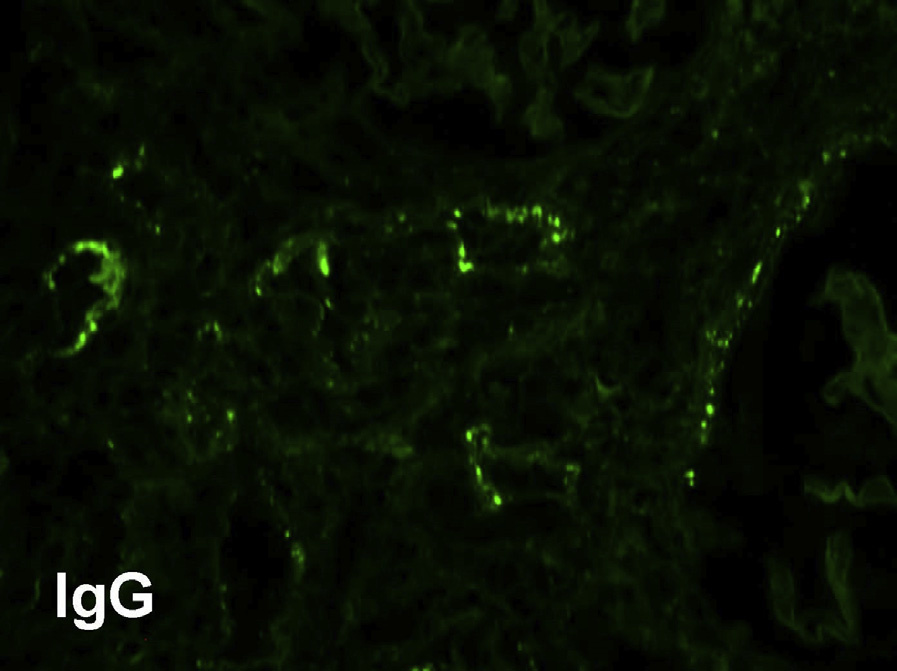
Immunofluorescence microscopy: Granular tubular basement membrane immune deposits staining for IgG (IgG4 and also IgG1 and IgG3) and C3 and sometimes C1q occur in >80% of biopsies. The deposits are typically limited to areas with inflammation. Biopsies with MN show frequent granular peripheral glomerular capillary wall deposits for IgG (usually, but not always IgG4 dominant), which do not stain for PLA2R, the most commonly identified antigen in cases of primary MN. Mesangial deposits may be present. Those with Henoch-Schönlein purpura show IgA deposits.
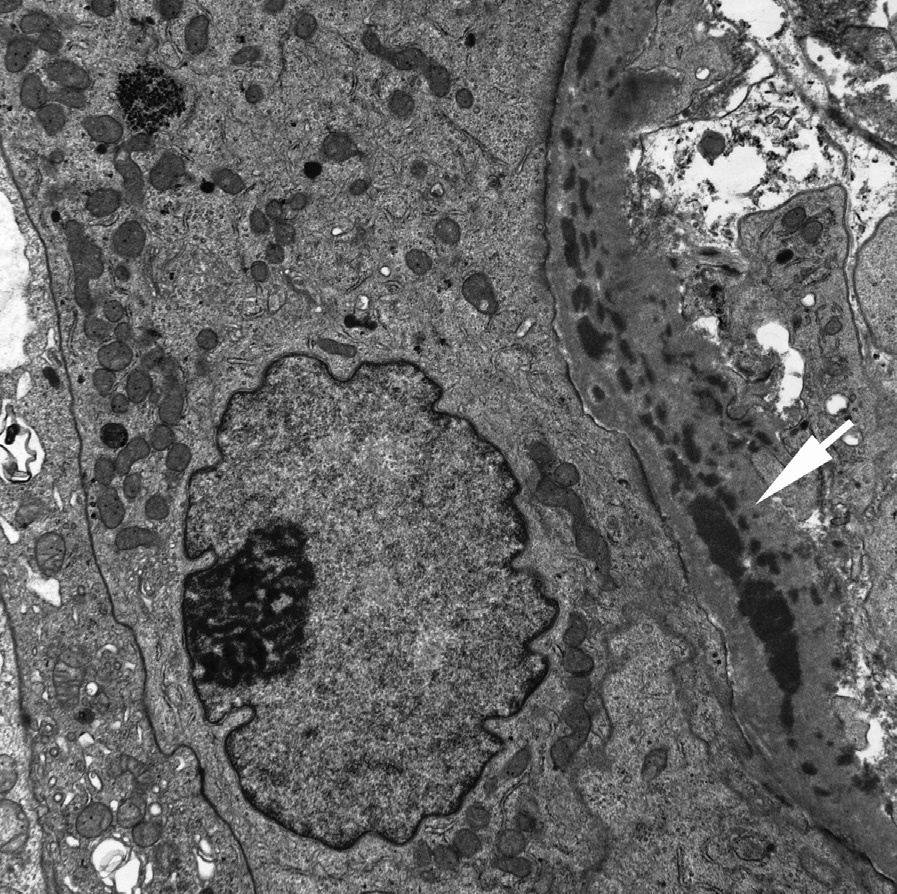
Electron microscopy: Immune-type tubular basement membrane deposits consistent with immunofluorescent findings are commonly identified. Abundant glomerular subepithelial deposits are present in those with MN. Mesangial and subendothelial deposits may be present.
The etiology of IgG4-related disease is poorly understood. Autoimmunity, hypersensitivity, and innate immunity might be involved. It is not clear if the elevated IgG4 is pathogenic or a secondary phenomenon. CD41 cytotoxic T cells are abundant and IL-4, IL-10, and TGF-β are increased in affected organs and may be involved in pathogenesis.
ANCA-associated vasculitis, especially granulomatosis with polyangiitis and eosinophilic polyangiitis (Churg-Strauss syndrome), may have abundant IgG4-positive plasma cells. Serum IgG4 may also sometimes be elevated in ANCA vasculitis. About 15% of TIN of other causes contain IgG4-positive plasma cells. Other conditions that may have abundant IgG4-positive plasma cells include diabetic nephropathy, lupus nephritis, MN, and idiopathic TIN.
Necrotizing angiitis, granulomatous lesions, neutrophil infiltration, and advanced tubulitis are very rare in IgG4-related TIN, and if present, largely rule out this condition. Multicentric Castleman disease, Erdheim-Chester disease, and lymphoproliferative disorders may be associated with dense plasmacytic infiltrate and similar systemic manifestations.