Microscopy Images
Light chain deposition disease (LCDD) is the most common of the non-AL amyloid monoclonal immunoglobulin deposition diseases (MIDD), which also include subtypes with both light and heavy chains or only heavy chain component. The specific type of MIDD is diagnosed by immunofluorescence or immunohistochemistry.
Patients with LCDD are middle-aged or older adults, and men are affected more often than women. The kidney is the major organ involved, often with nephrotic range proteinuria. Patients less commonly have dominant tubular basement membrane, with lesser glomerular deposits and little or no proteinuria. Patients may also show signs related to the underlying plasma cell dyscrasia, such as anemia.
Other organs may be involved, such as the liver or heart, but these extrarenal deposits do not usually cause clinical signs. Some patients have concomitant light chain cast nephropathy caused by the monoclonal protein, with resulting acute kidney injury. Patients who are eligible for treatment of the underlying plasma cell dyscrasia/multiple myeloma have hematologic response in about half of cases, with corresponding stabilization or even improvement of end-organ function. Most untreated patients develop end-stage renal disease. Patients who undergo kidney transplantation despite persistent monoclonal protein develop recurrent disease in the transplant.
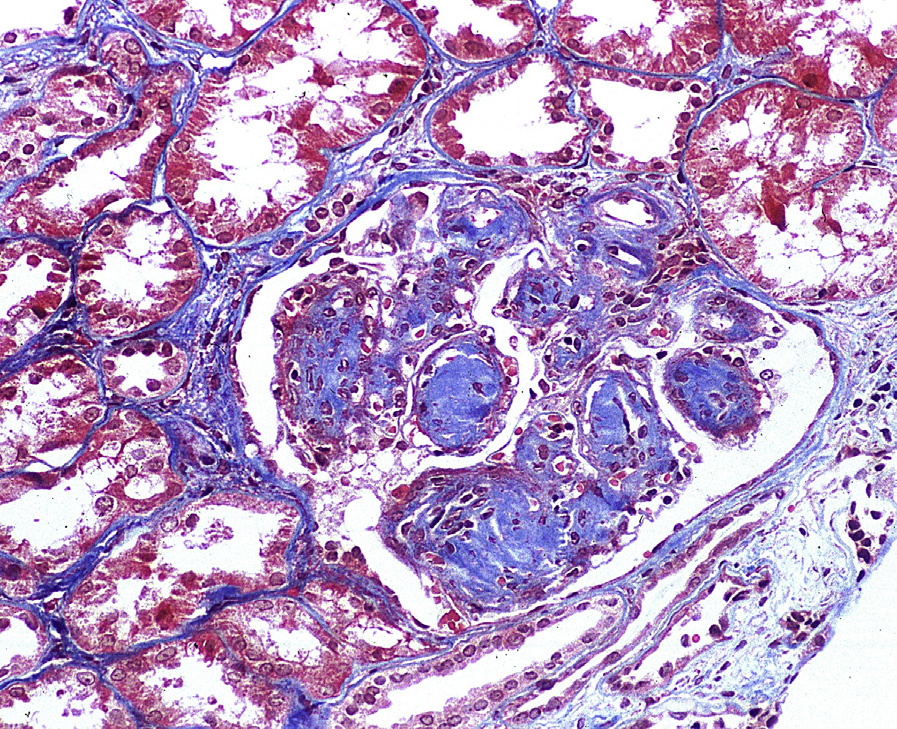
Light microscopy: Advanced LCDD shows nodular sclerosis, with very similar appearance to diabetic nephropathy by light microscopy. Early LCDD shows only mild mesangial expansion. Congo Red stain is negative.
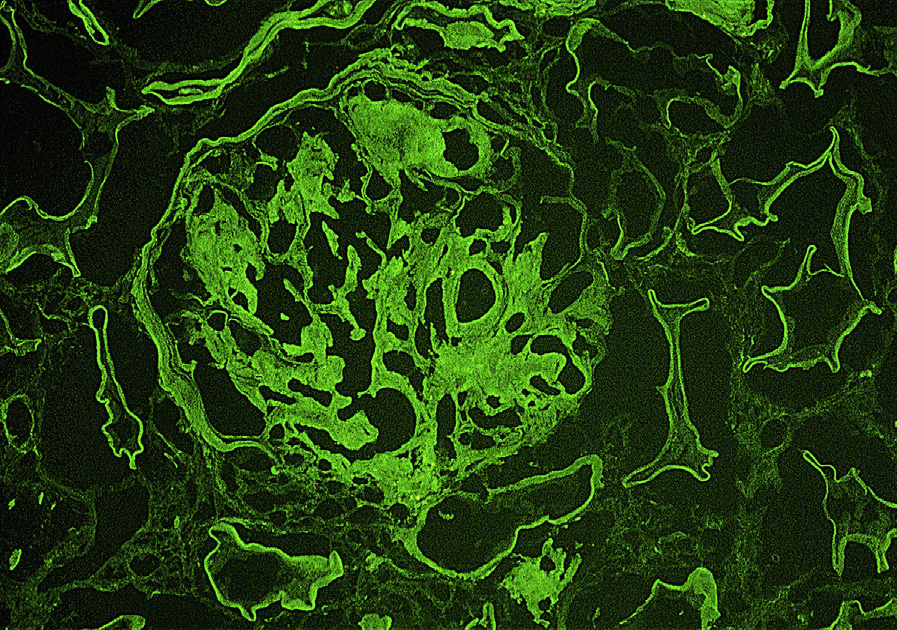
Immunofluorescence microscopy: Monoclonal light chain staining, most commonly κ light chain, in mesangium and along glomerular and tubular basement membranes.
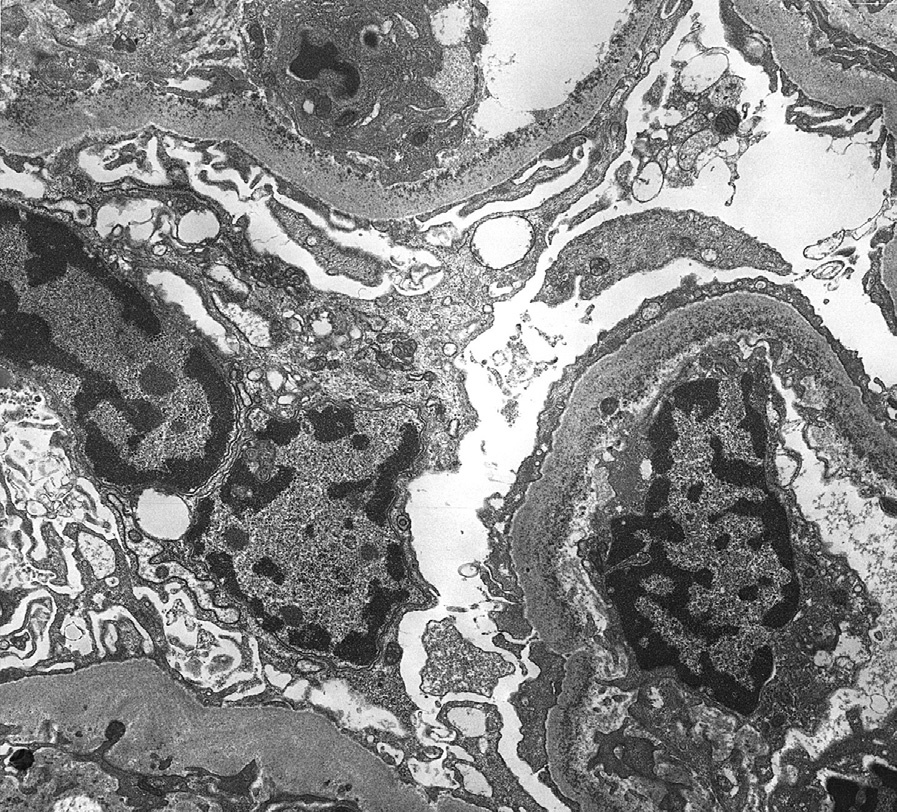
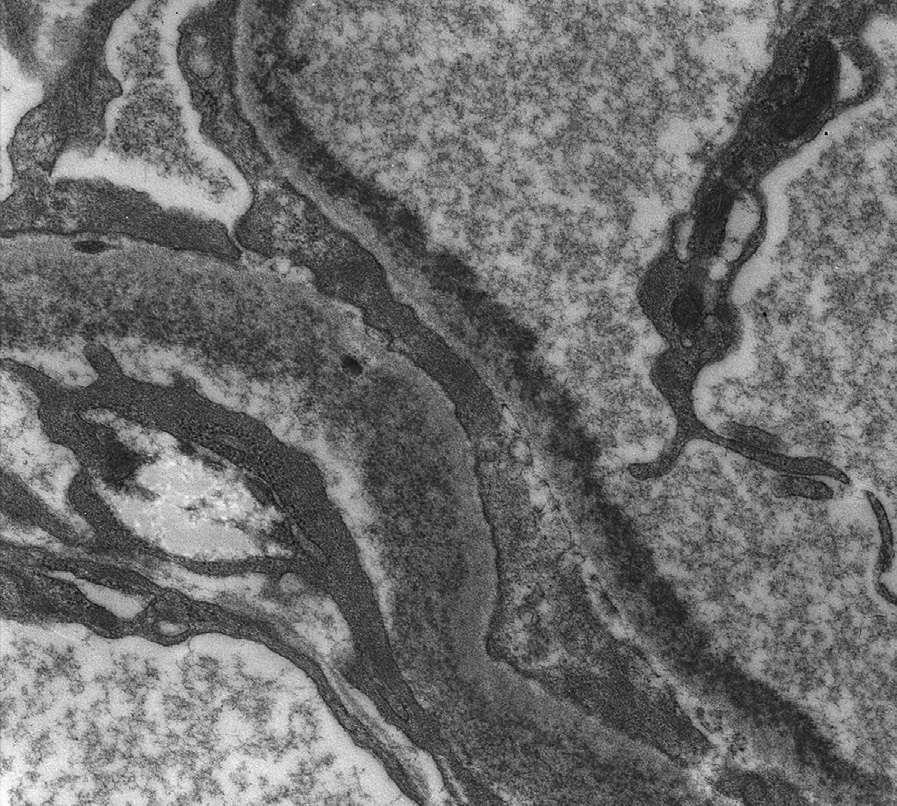
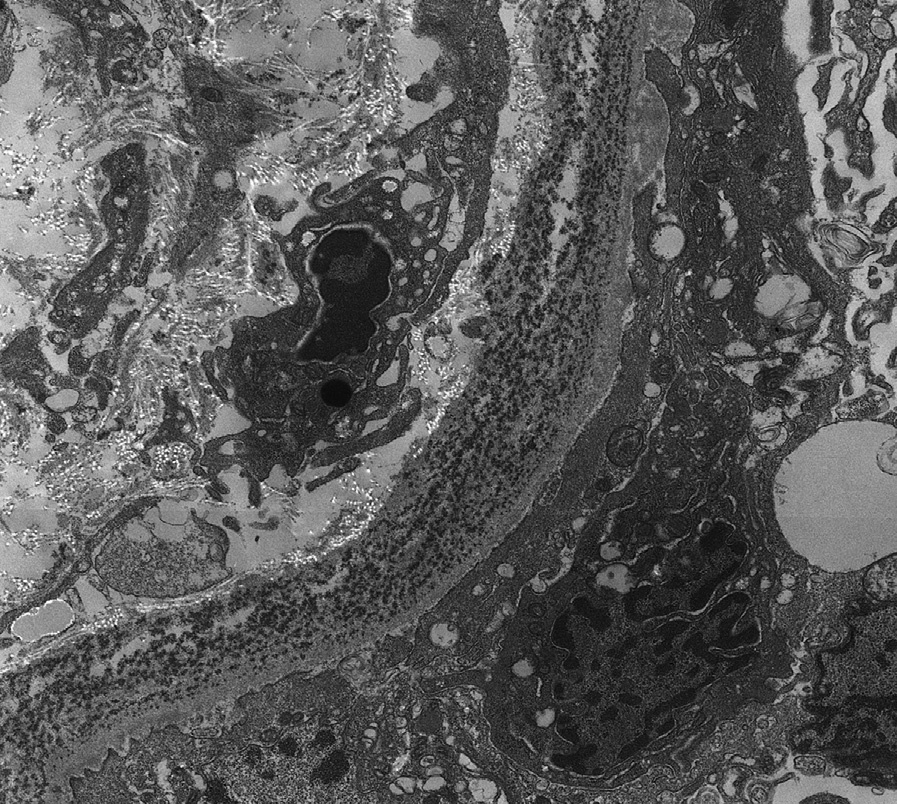
Electron microscopy: Mesangial and inner glomerular and outer tubular basement membrane deposits of extracellular punctate, powdery, ground-pepper–like deposits, with extensive foot process effacement. In some early cases, deposits may only be detected by immunofluorescence.
LCDD is due to deposition of a monoclonal light chain in mesangial areas and along glomerular and tubular basement membranes. The monoclonal protein is due to underlying plasma cell dyscrasia. About half of patients have multiple myeloma by bone marrow examination. Patients with LCDD without diagnostic multiple myeloma were previously said to have monoclonal gammopathy of undetermined significance, now called monoclonal gammopathy of renal significance.
LCDD may be distinguished from other causes of nodular sclerosis and mesangial expansion by specific light chain staining in glomeruli and tubular basement membranes, negative Congo Red stain, and punctate amorphous, ground-pepper–like appearance of deposits on electron microscopy.
Fibrillary glomerulonephritis is Congo Red negative, and has polyclonal immunoglobulin G and proliferative appearance; diabetic nephropathy shows no deposits; and other MIDD show light and heavy chain staining or only heavy chain staining. Other causes of a membranoproliferative glomerulonephritis pattern show disease-specific immunofluorescence and electron microscopic appearances.