Microscopy Images
Hyperoxaluria may be due to hereditary defects of key enzymes of oxalate metabolism, or secondary to excess intake or increased enteric absorption. Inherited forms are autosomal recessive; the most common, type 1, accounts for ~80% of cases and presents in childhood with recurrent nephrolithiasis. About half of patients with childhood onset of nephrolithiasis develop end-stage kidney disease by age 15, with accompanying systemic oxalate deposits, including cardiovascular system, eyes, and bones. Kidney disease is milder with type 2 or type 3 hyperoxaluria.
Enteric hyperoxaluria occurs in malabsorption syndromes (eg, after bariatric surgery) and with pancreatic disease or chronic inflammatory bowel disease.
High oxalate intake—from ingestion of vitamin C (which is metabolized to oxalate), spinach, rhubarb, beets, chocolate, black tea, or ethylene glycol—can also cause oxalosis. Calcium oxalate crystals within tubules cause acute tubular injury, resulting in acute kidney injury, with or without nephrolithiasis. If the underlying cause can be corrected, recovery is possible. Pyridoxine, a cofactor for aminotransferase, may be of benefit in some patients with type 1 primary hyperoxaluria, as it may enhance residual enzyme activity.
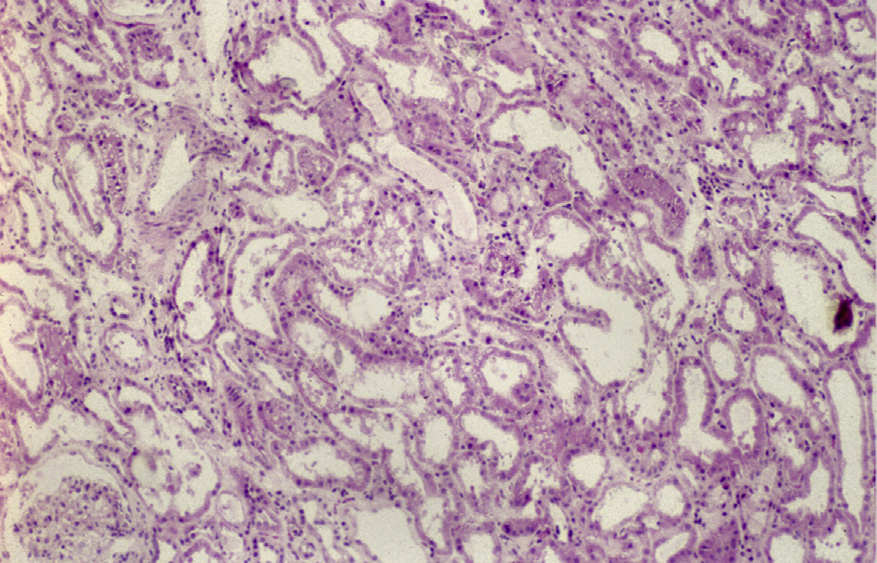
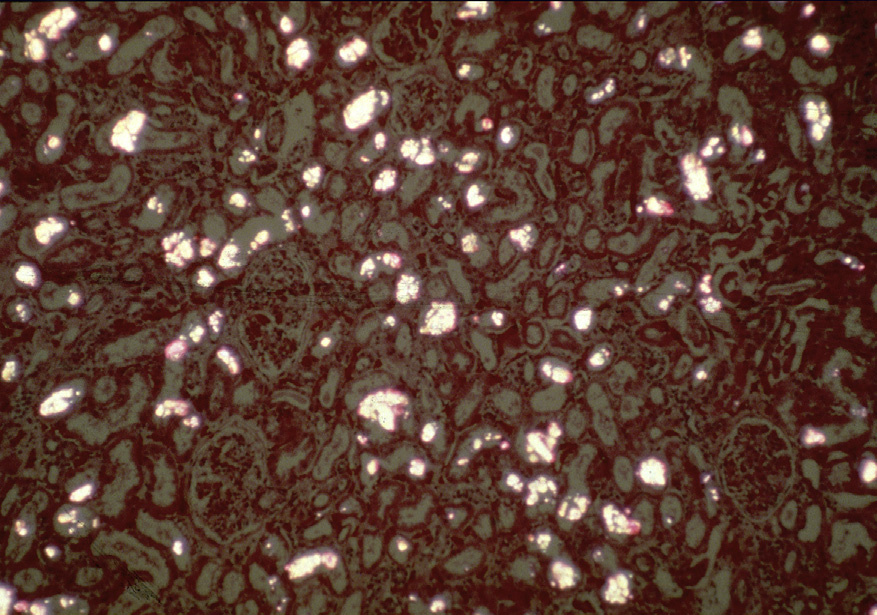
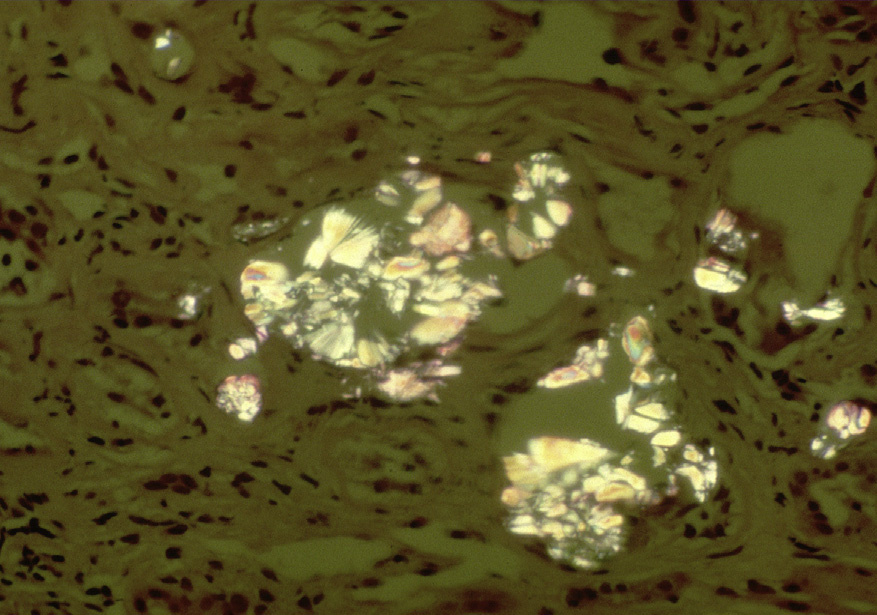
Light microscopy: There is acute tubular injury, with sloughing, vacuolization of proximal tubular cells, and flattened, simplified epithelium with frequent intratubular crystals, which are clear on hematoxylin and eosin stain under standard light microscopy and show multicolored birefringence under polarized light, with classic fan-shaped morphology.
Calcium oxalate crystals are not typically visible by periodic acid–Schiff, Jones, or Masson trichrome stains, as they are removed during processing.
Immunofluorescence microscopy: No staining.
Electron microscopy: No deposits or other specific abnormalities. Calcium oxalate crystals are visible and polarizable on toluidine blue–stained semi-thick sections.
Oxalosis results from supersaturation of the urine due to excess oxalate in the presence of calcium, resulting in calcium oxalate crystals. Numerous etiologies exist. Type 1 hyperoxaluria, the most common of the hereditary defects, is due to, the deficiency of hepatic microsomal alanine glyoxylate aminotransferase, with an incidence of 1:120,000 and autosomal recessive inheritance. Pyridoxine is a cofactor for this enzyme. Among children with nephrocalcinosis and/or urolithiasis, nearly 7% have this underlying defect.
Accounting for 10% of primary hyperoxaluria cases, type 2 is due to deficiency of glyoxylate reductase/ hydroxypyruvate reductase, resulting in increased oxalate and L-glyceric acid, usually with less severe kidney disease than in type 1. Type 3 is due to loss of function of mitochondrial 4-hydroxy-2-oxoglutarate aldolase, with resulting aberrant hydroxyproline metabolism and increased oxalate.
Enteric hyperoxaluria occurs due to nonabsorbed fatty acids binding calcium in the proximal intestine, resulting in less calcium available to bind oxalate. Thus, excess soluble oxalate is absorbed from the intestinal tract and excreted by the kidney, where calcium oxalate crystals form. Ethylene glycol and vitamin C are metabolized to oxalate, and can also cause oxalosis.
Oxalate crystals cause mechanical tubular obstruction, resulting in acute tubular injury. Phagolysosomes are also overloaded by indigestible nanocrystals, causing cell death and inflammation. A specific type of cell death, necroptosis, is caused by crystals activating receptor-interacting protein kinase 3–mediated phosphorylation of the pseudokinase mixed lineage kinase domain-like. Necroptosis of parenchymal and inflammatory cells results in release of large amounts of histones that may be directly cytotoxic, amplifying injury.
Damage-associated molecular patterns (DAMPs) further activate inflammatory mechanisms, collectively termed the inflammasome.
Rare or occasional calcium oxalate crystals can be seen in acute or chronic tubular injury, especially in kidney transplant biopsies, as a nonspecific phenomenon, while in oxalate nephropathy there are frequent crystals associated with tubular epithelial cell injury.
Crystals due to 2-8-hydroxyadenine show similar appearance to oxalate under polarized light, but stain brown with H&E. Calcium phosphate crystals are round, globular, bluish-purple and nonpolarizable.