Microscopy Images
Sickle cell disease is an autosomal recessive disease that occurs most commonly in individuals of African ancestry, with an incidence of 1 in 500 African Americans. Kidney disease has varying manifestations, with microalbuminuria in childhood, progressing to overt proteinuria in 20%-25% and nephrotic syndrome in about 5%, and progressive GFR loss after age 20 years in 5%-8% of all sickle cell patients. Patients often show hyposthenuria and hematuria as well. Some patients develop papillary necrosis and acute kidney injury due to sickle cell crisis. Patients with either homozygous sickle cell disease or sickle cell thalassemia can develop any of these manifestations related to sickling. Sickle cell trait has been reported to cause sickle cell nephropathy only in rare cases.
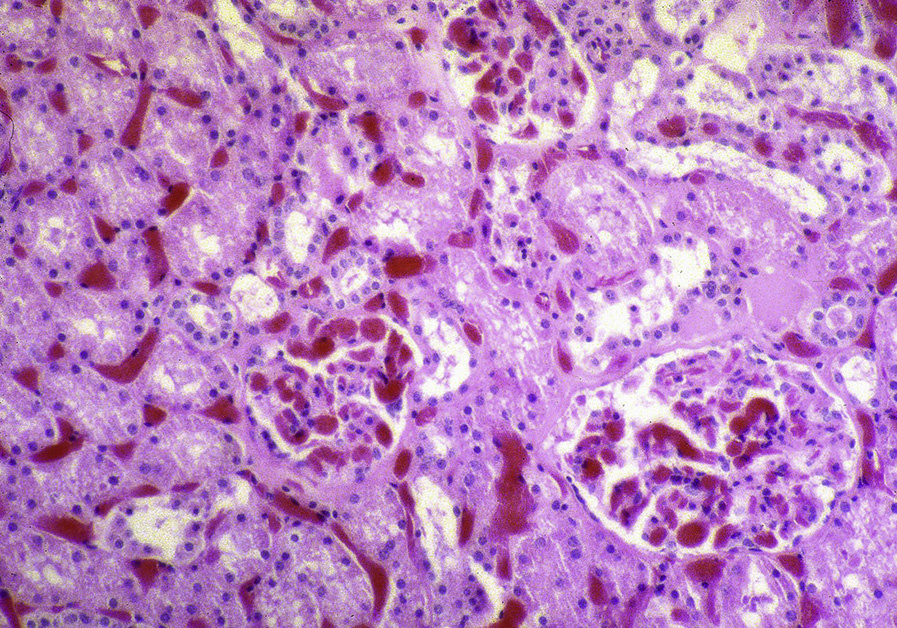
Light microscopy: Cortical infarcts can occur in sickle cell crisis due to occlusion of arteries. With chronic injury, glomerulomegaly and global and secondary segmental glomerulosclerosis are present, often in a perihilar location, with glomerular basement membrane contour duplication and corrugation.
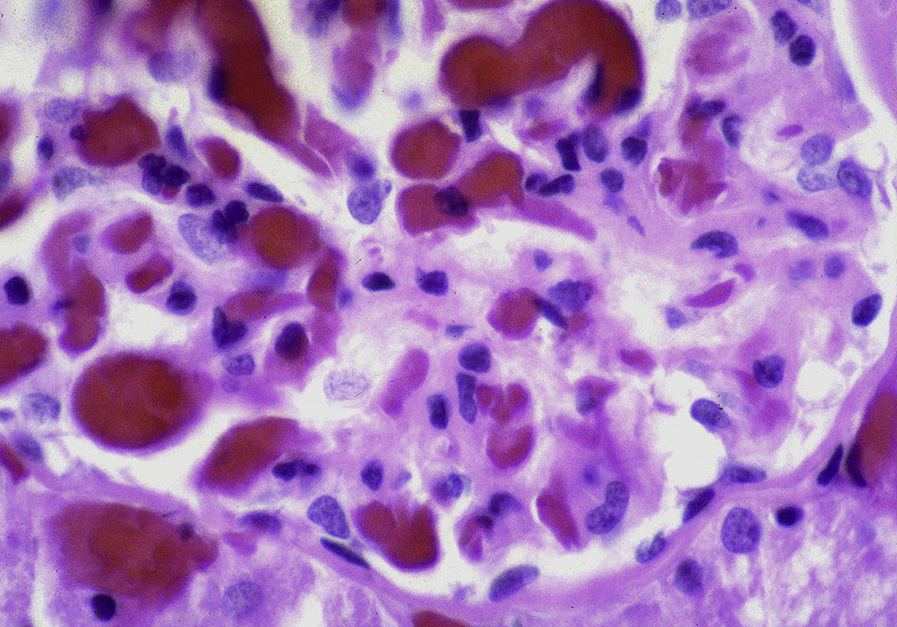
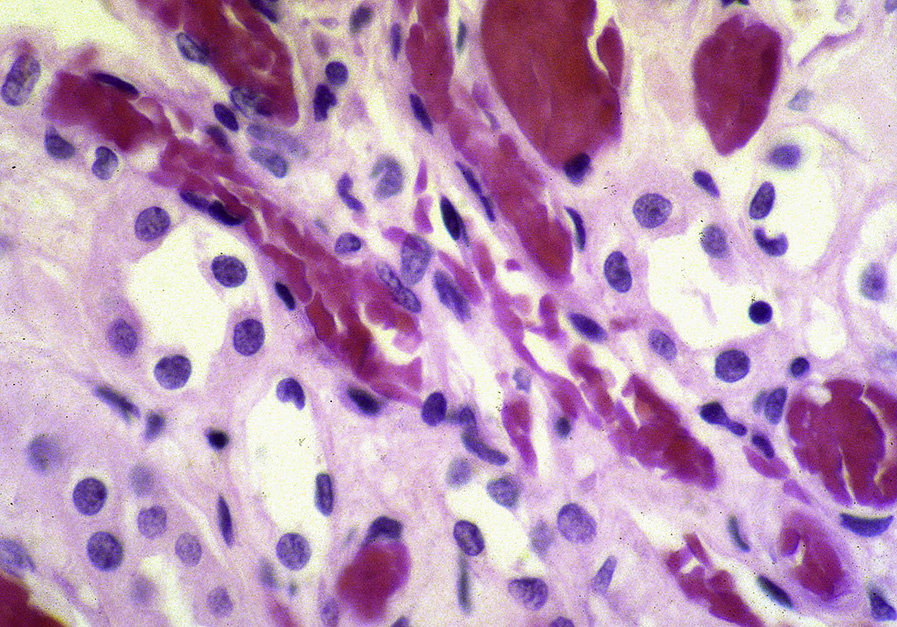
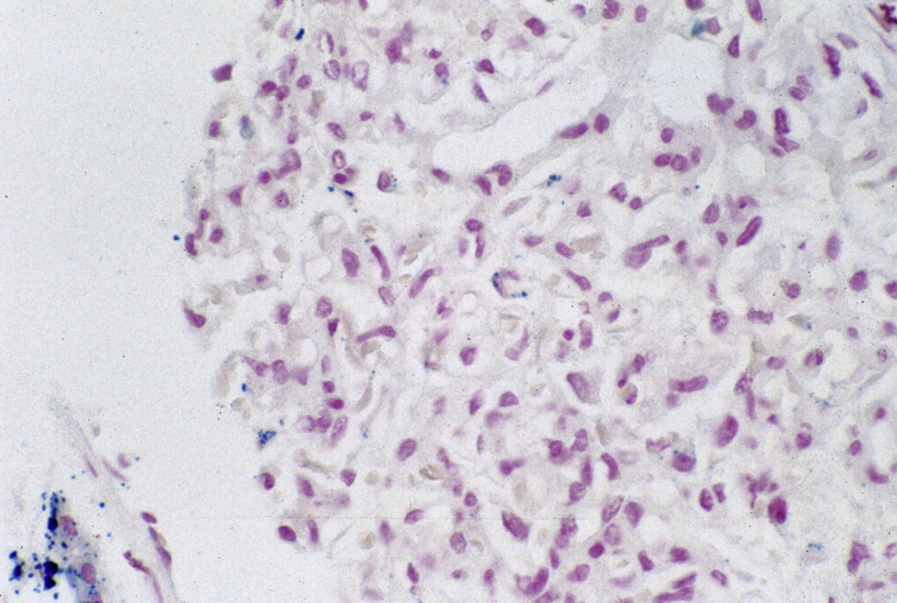
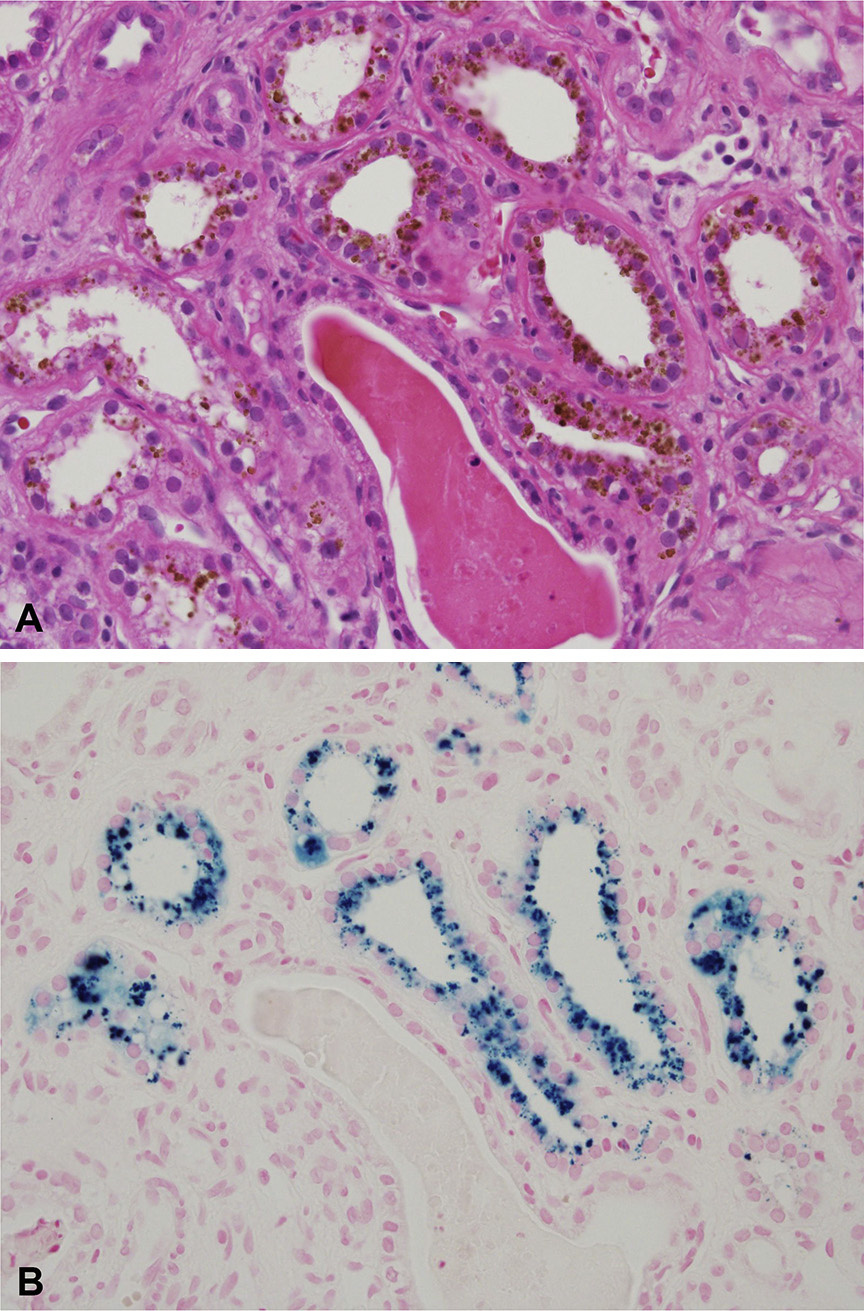
Collapsing lesions may be present related to vascular compromise resulting in ischemia. There is prominent hemosiderin deposition, seen as reddish granules in tubular epithelial cells and staining positive on Prussian blue iron stain, with focal deposition in glomerular epithelial cells. Sickled red blood cells are present, most often in medullary peritubular areas and more rarely in glomeruli. Interstitial fibrosis and tubular atrophy are present to a varying extent.
Immunofluorescence microscopy: Nonspecific IgM and C3 staining in scarred glomeruli.
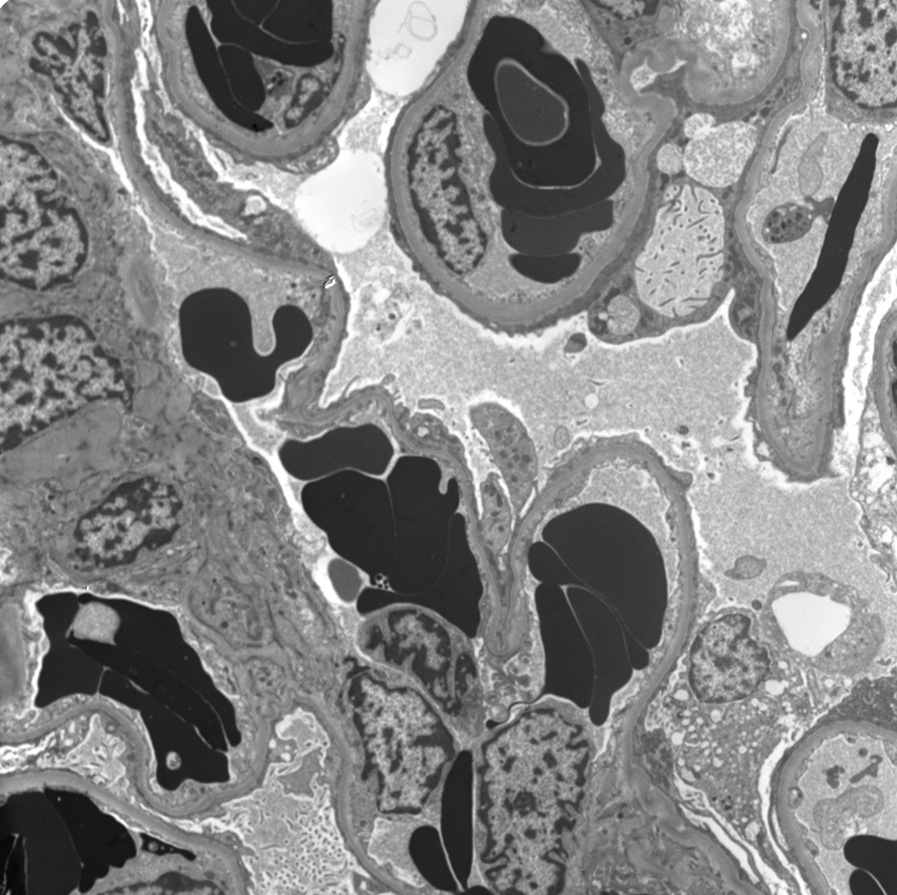
Electron microscopy: Sickle-shaped red blood cells in glomerular and peritubular capillaries with polymerized hemoglobin.
Glomerular basement membranes show corrugation and expansion of lamina rara interna and cellular interposition without immune deposits, with subtotal foot process effacement in glomeruli without sclerosis.
Sickle cell disease is due to a single-base mutation that results in a single amino acid substitution (valine for glutamate) in hemoglobin, and causes hemoglobin polymerization and sickling under low oxygen tension, particularly with volume depletion and exercise.
Sickling triggers sickle cell crisis due to occlusion of vessels. The vasa recta are most susceptible due to relative hypoxia, low pH, and hypertonicity in the medulla. Secondary segmental sclerosis with gradual development of proteinuria and CKD is postulated to be due to chronic hypoxia, endothelial injury related to iron/heme and sickling of red blood cells, nitric oxide imbalance, and altered hemodynamics, related to marked glomerular hypertrophy/hypertension and hyperfiltration.
Primary focal segmental glomerulosclerosis presents with nephrotic syndrome without hemosiderin deposition, and shows extensive foot process effacement even in nonsclerotic glomeruli. Chronic thrombotic microangiopathy has glomerular basement membrane double contours, however, it lacks congestion and sickled cells within peritubular and glomerular capillaries. Hemolysis results in pigmented granular casts that show positivity on Prussian blue iron stain, but an absence of sickled cells within capillaries.