Microscopy Images
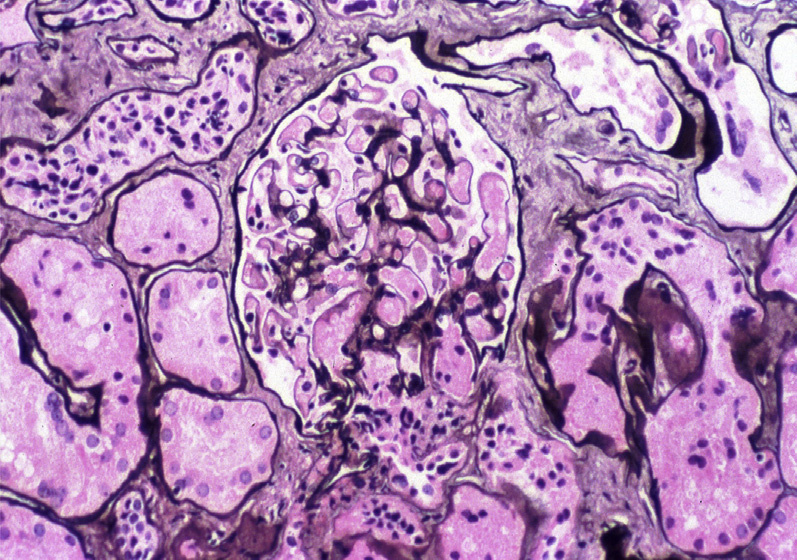
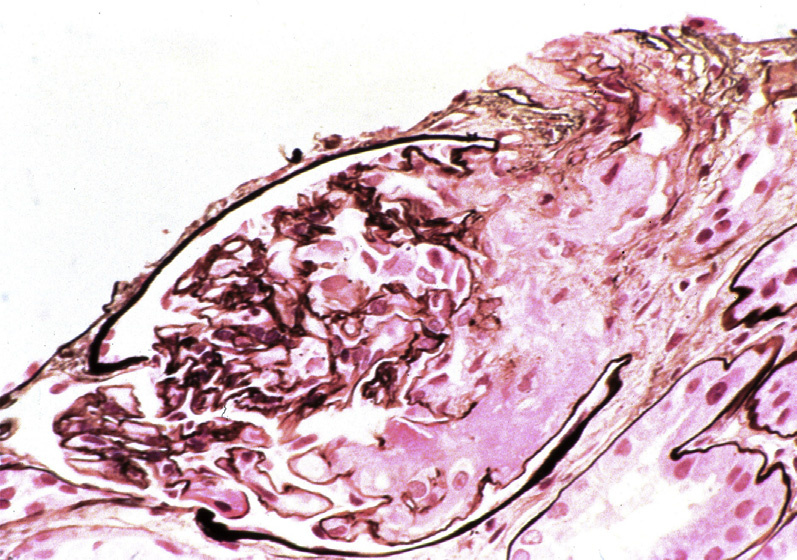
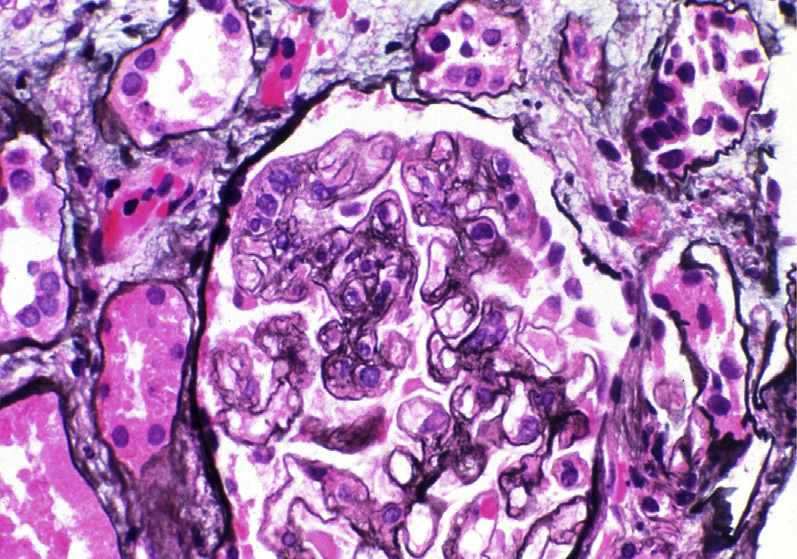
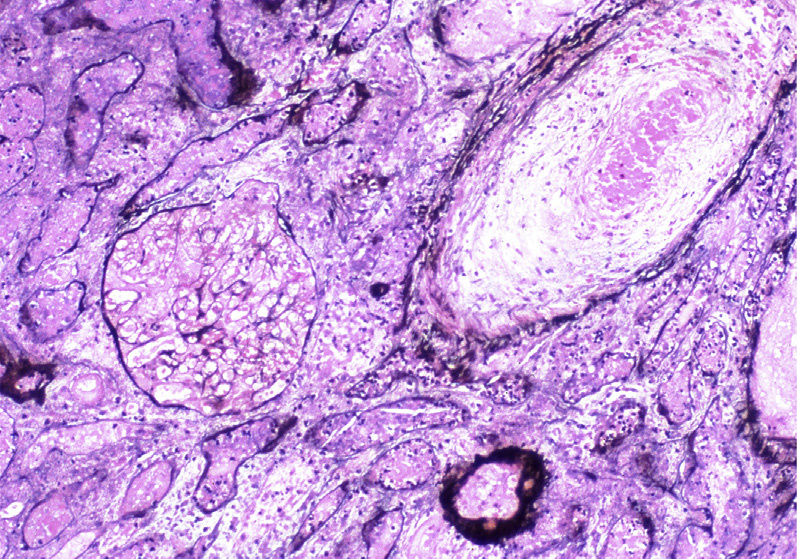
Thrombotic microangiopathy (TMA) is a lesion with multiple etiologies. The presentation depends on the cause, and typically includes the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. Hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) both manifest as TMA. In addition to classic TMA findings, HUS typically presents with bloody diarrhea, fever, and hypertension. TTP typically presents with fever, hypertension, mild proteinuria, and neurologic symptoms. TMA may occur at any age, but HUS due to E. coli is more common in children.
Light microscopy: In HUS, there are fibrin thrombi (often with fragmented red blood cells) within glomeruli, in subendothelial areas, and in the mesangium. These are accompanied by mesangiolysis, endothelial swelling, corrugation of the glomerular basement membrane (GBM), and a bloodless appearance in segments of glomeruli uninvolved by thrombi. With causes of TMA other than HUS, arterioles and arteries are more often involved.
Arterioles and arteries can have intimal proliferation with mucoid changes and entrapped red blood cell fragments, or frank necrosis and/or fibrin thrombi.
Cortical necrosis can be seen in severe cases.
Chronic changes include duplication of GBMs resulting in a membranoproliferative-like pattern, and focal segmental and global glomerulosclerosis with proportional tubulointerstitial fibrosis. Intimal proliferation and fibrosis with narrowed lumens of arterioles/interlobular arteries give rise to the socalled onion-skin lesions. Recanalized thrombi may be present.
Immunofluorescence microscopy: Thrombi stain positive for fibrinogen. There may be nonspecific staining for IgM in glomeruli, and less frequently C3 and IgG.
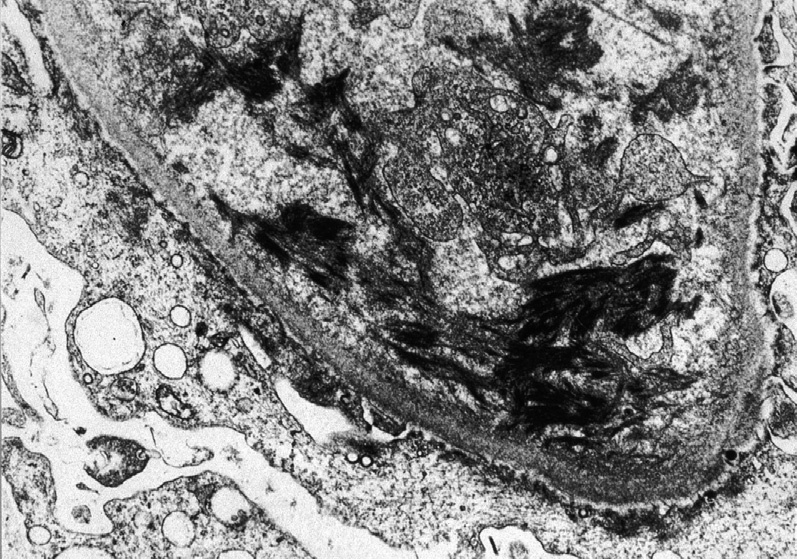
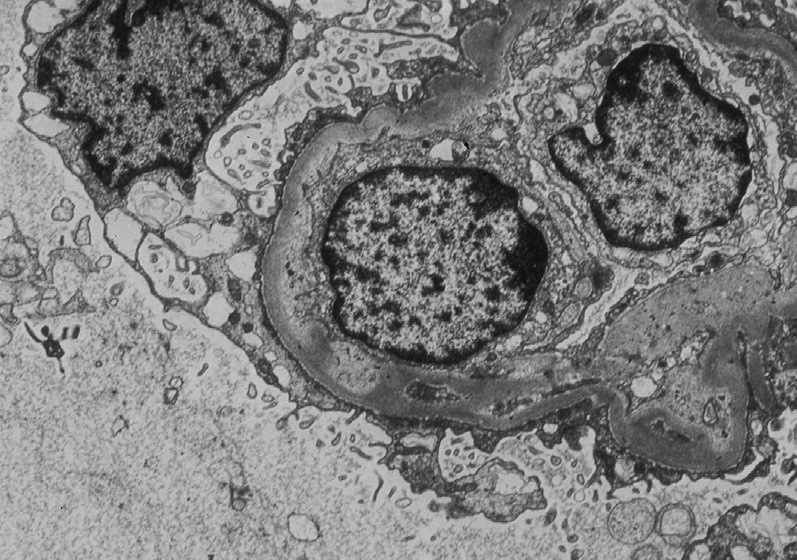
Electron microscopy: There are swollen glomerular endothelial cells with loss of fenestrations, expansion of lamina rara interna, mesangiolysis, and fibrin tactoids with platelets and fragmented red blood cells.
Chronic findings include interposed cells with new GBM matrix material deposition.
HUS is predominantly related to endothelial injury and activation of a secondary prothrombotic state.
Typical HUS is caused by enteric pathogens with Shiga-like and Shiga toxins. TTP is predominantly related to deficiency of or acquired autoantibodies against ADAMTS13.
Other causes of TMA include complement dysregulation (atypical HUS), various drugs, scleroderma, malignant hypertension, eclampsia/preeclampsia, and antibody-mediated rejection.
Thrombotic microangiopathy has similar morphologic findings regardless of the underlying etiology. However, HUS/TTP predominantly affect glomeruli, whereas scleroderma and malignant hypertension predominantly affect arterioles and interlobular arteries. Transplant glomerulopathy in chronic antibody-mediated rejection has double contours of the GBM, often with donor-specific antibodies and C4d positivity in peritubular capillaries. Membranoproliferative pattern glomerulonephritis shows disease-specific staining by immunofluorescence and deposits by electron microscopy.